Docking
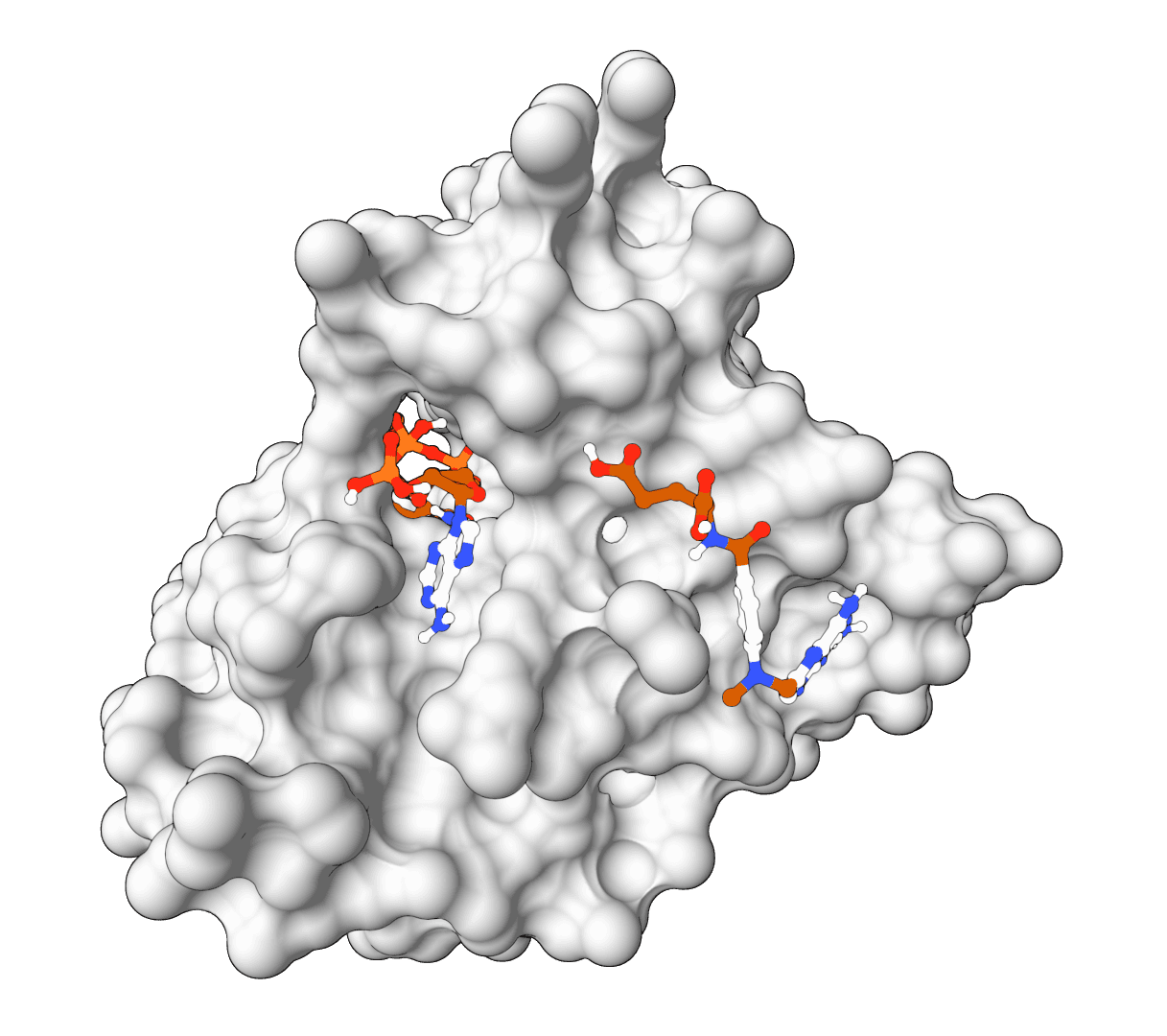
AutoDock Vina
AutoDock Vina is a widely-used molecular docking tool that predicts protein-ligand binding modes using physics-based force fields. Fast, reliable, and the gold standard for structure-based drug discovery.
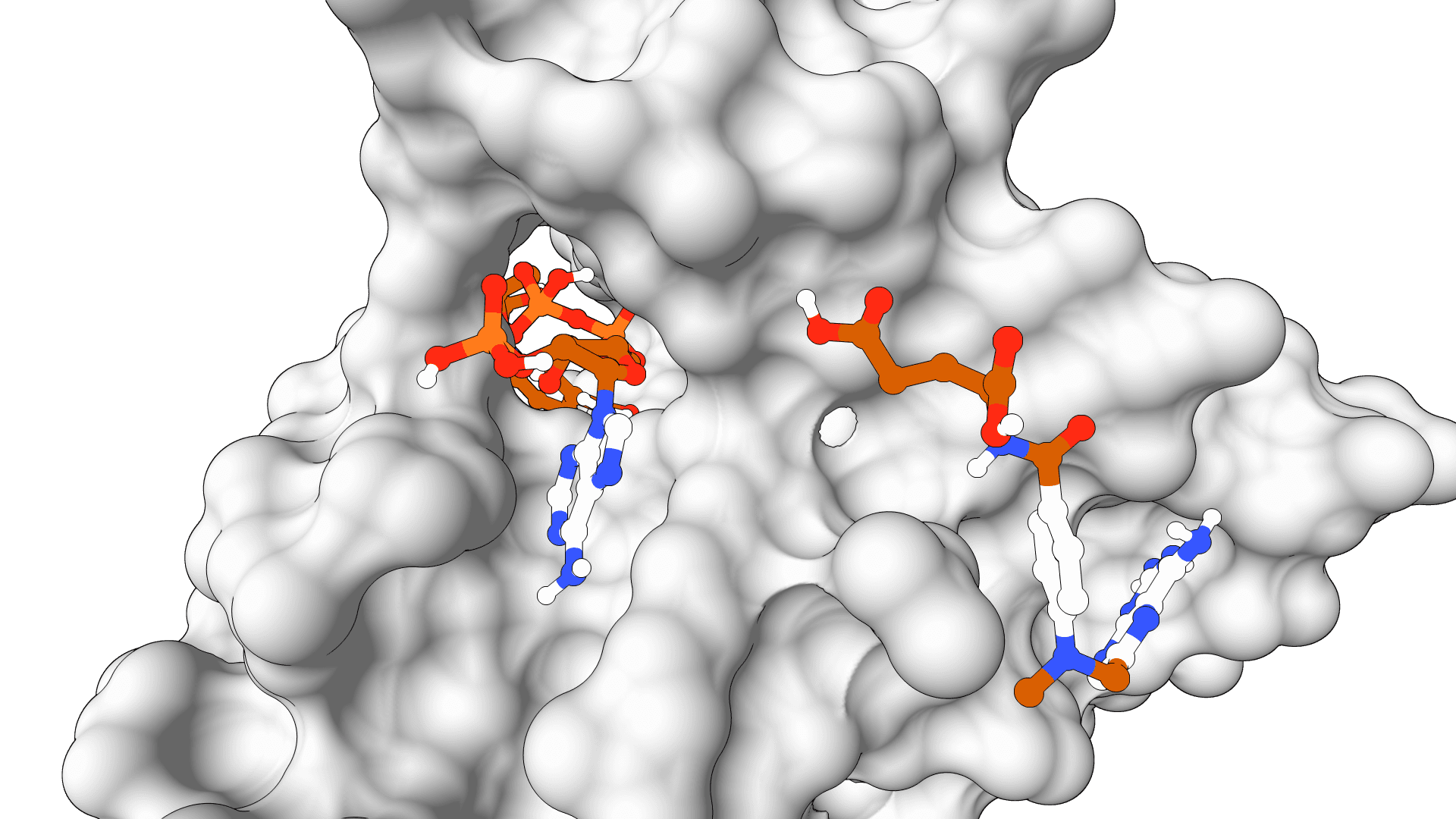
AutoDock-GPU
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
ColabDock
ColabDock is a protein-protein docking framework that uses AlphaFold2 to predict complex structures guided by experimental restraints from cross-linking mass spectrometry, NMR, or other sources.
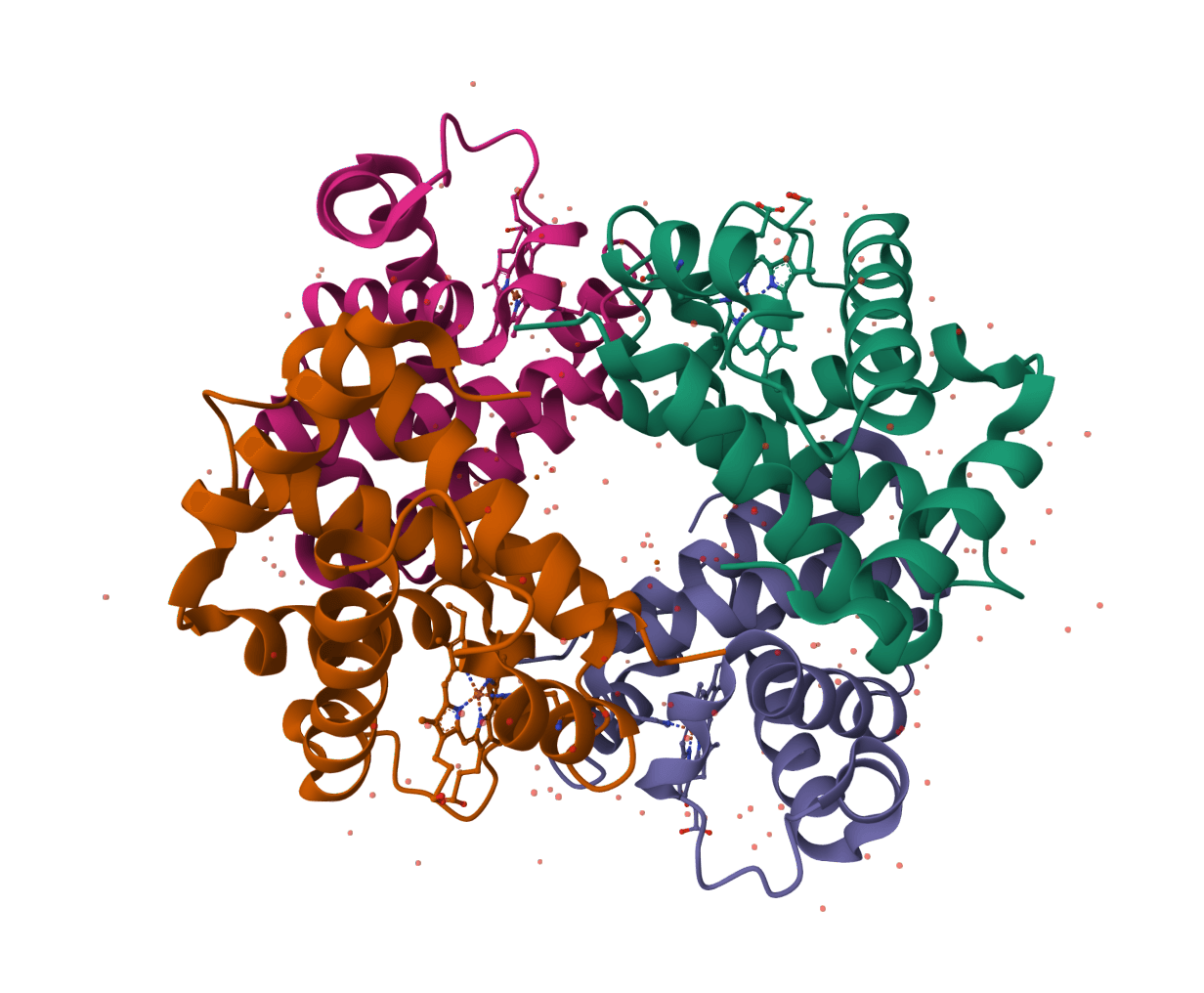
DFMDock
DFMDock (Denoising Force Matching Dock) is a diffusion model that unifies sampling and ranking for protein-protein docking within a single framework. It predicts docked poses for protein-protein complexes from unbound structures using denoising score matching with optional clash force guidance.
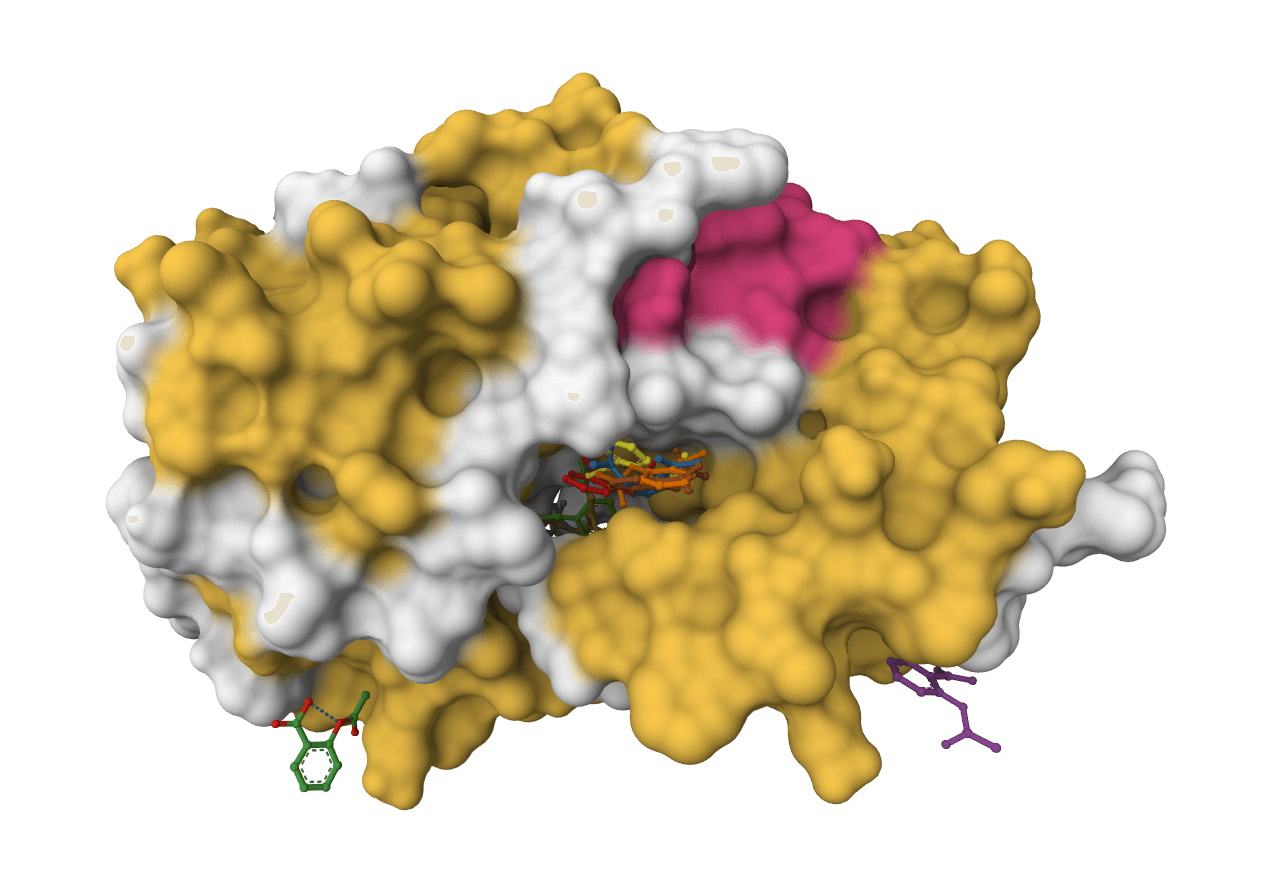
DiffDock-L
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
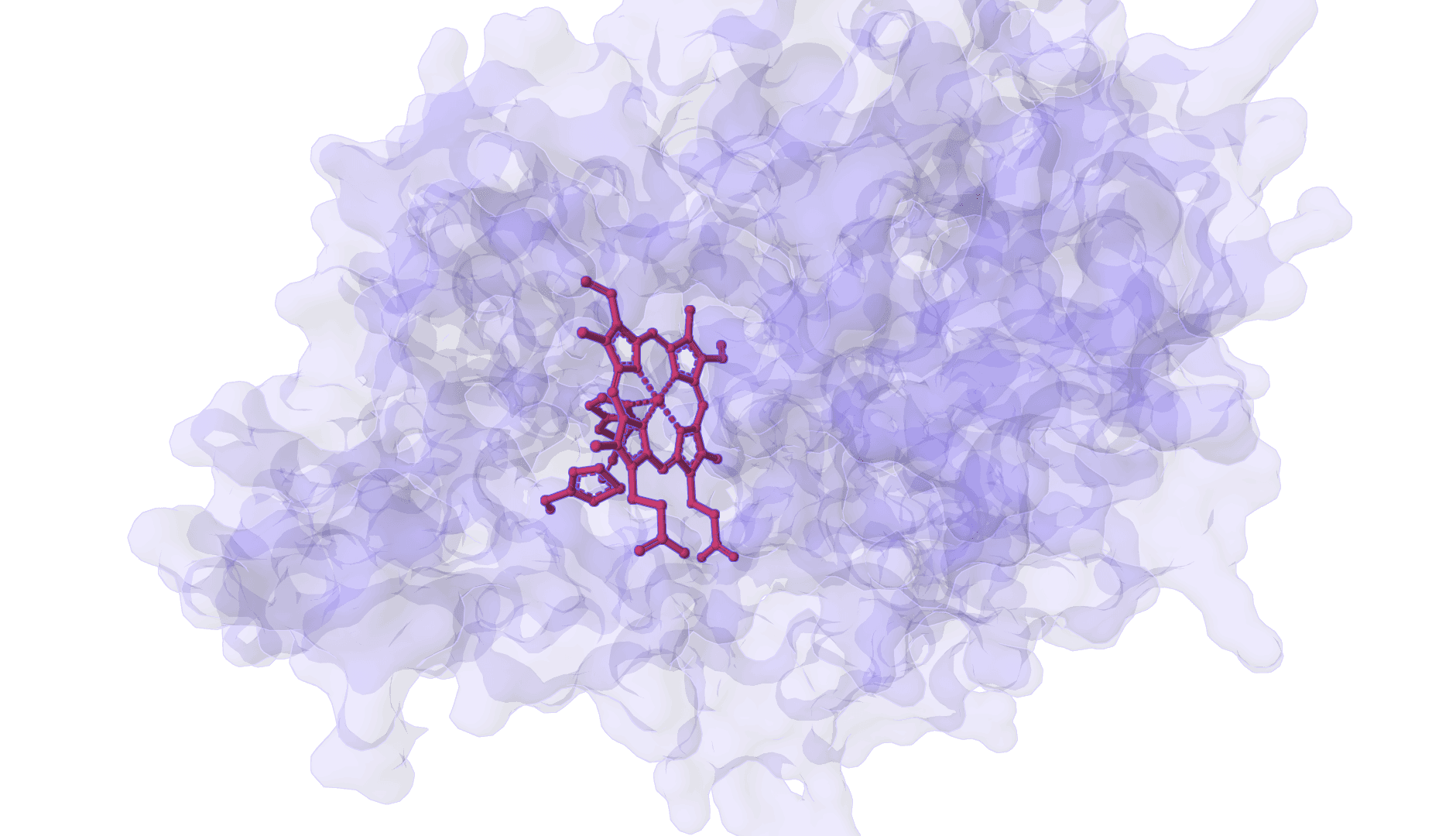
DynamicBind
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
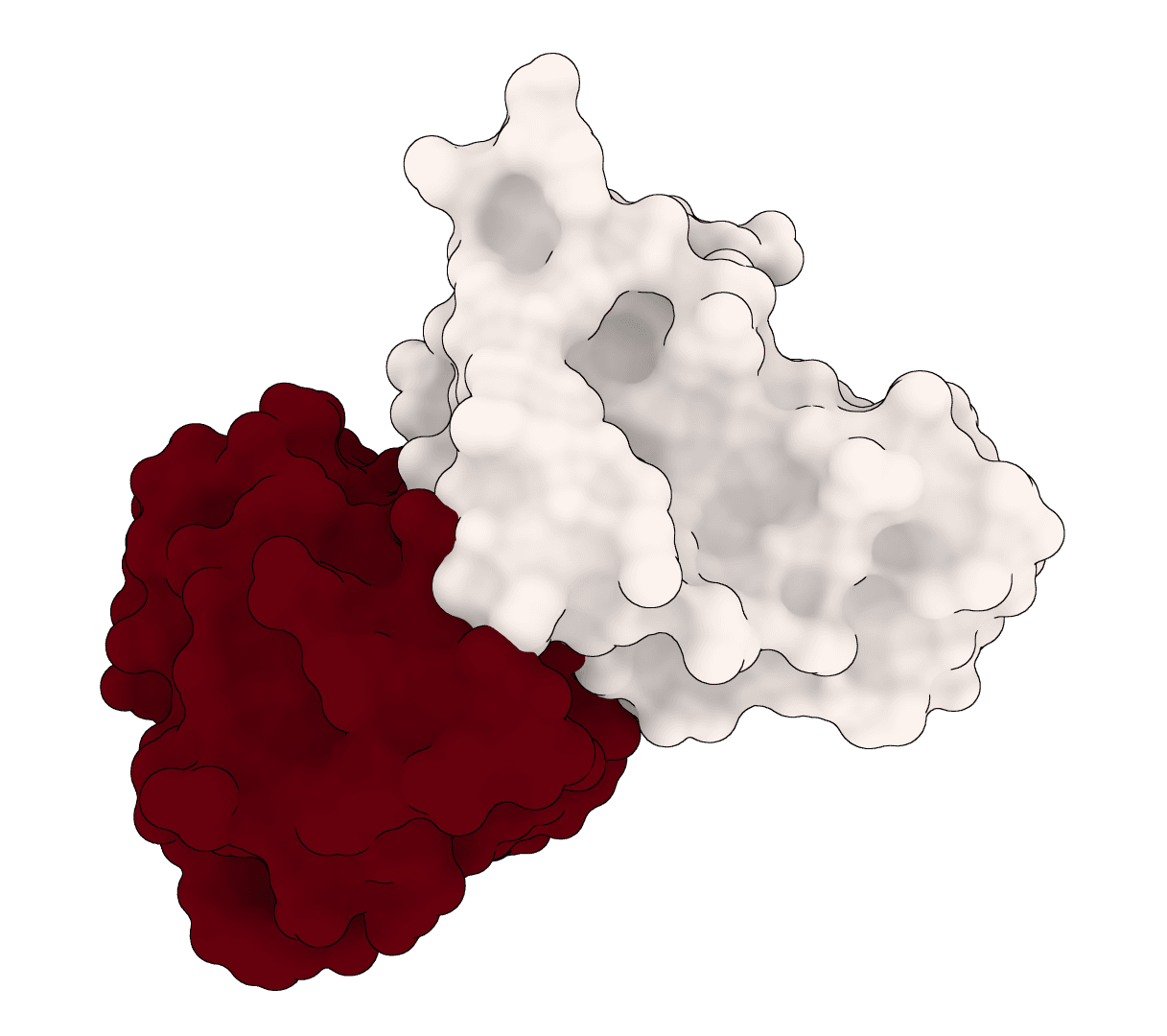
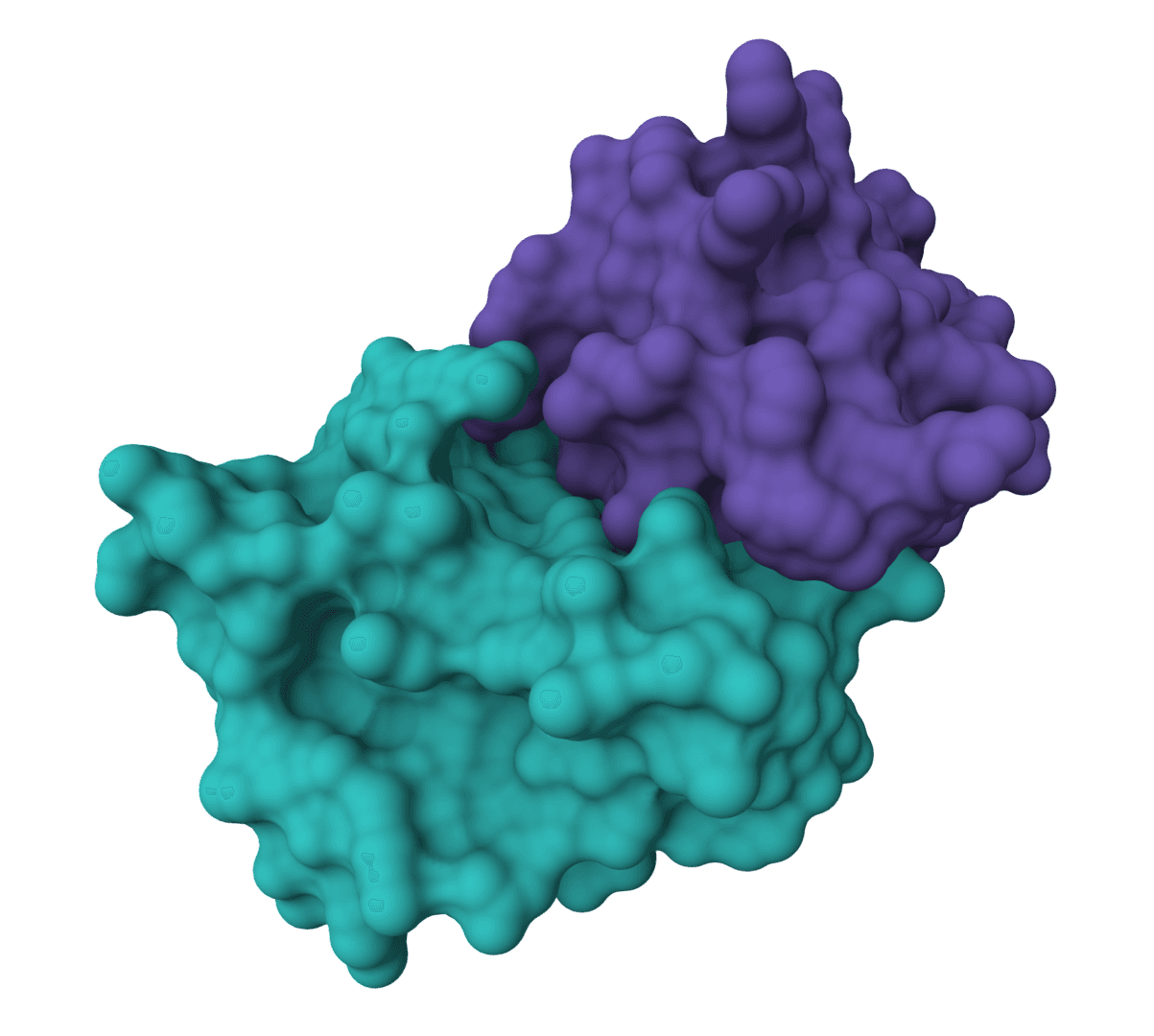
EquiDock
EquiDock is an SE(3)-equivariant graph neural network for rigid protein-protein docking. It predicts a binding pose for a protein-protein complex from unbound structures using geometric deep learning, with DIPS and DB5 pretrained checkpoints from the upstream release.
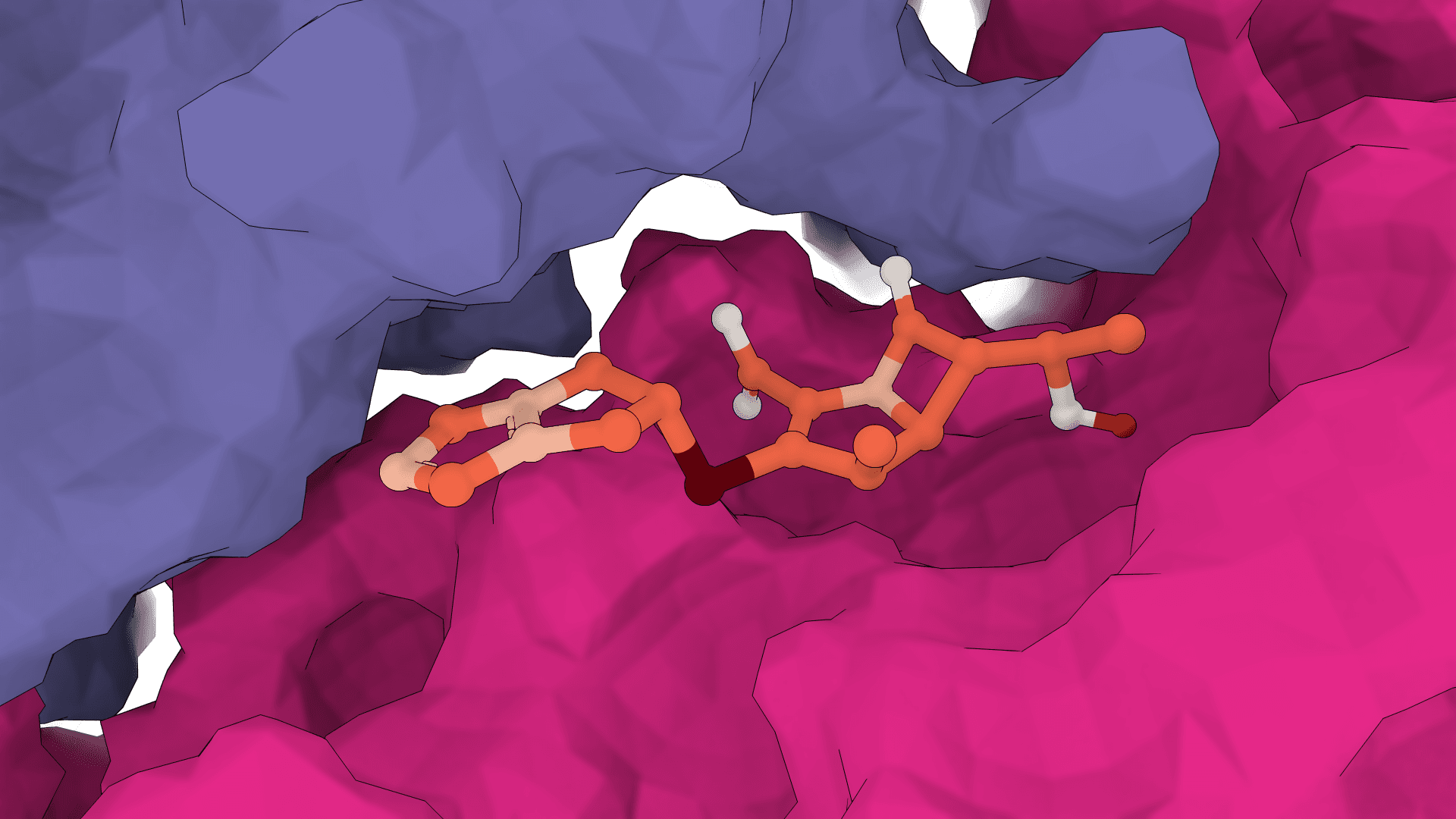
GNINA
GNINA is a molecular docking tool that combines traditional physics-based docking with deep learning CNN scoring for protein-small-molecule complexes. It provides accurate binding predictions with confidence scores, optimized for high-throughput virtual screening.
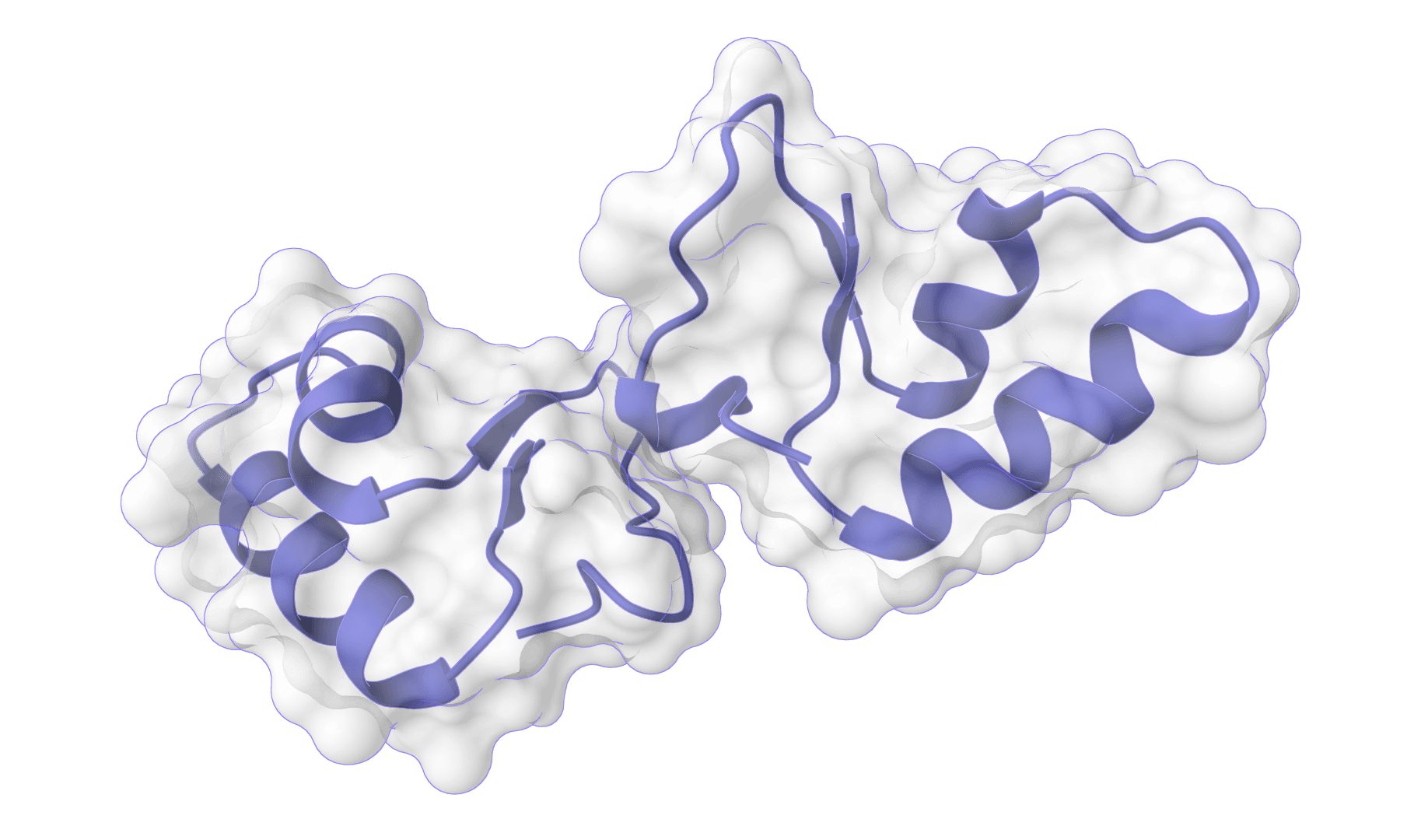
HADDOCK3
HADDOCK (High Ambiguity Driven protein-protein DOCKing) is an integrative modeling platform for biomolecular complexes. It uses experimental data and bioinformatic predictions to guide the docking process, generating accurate protein-protein complex structures.
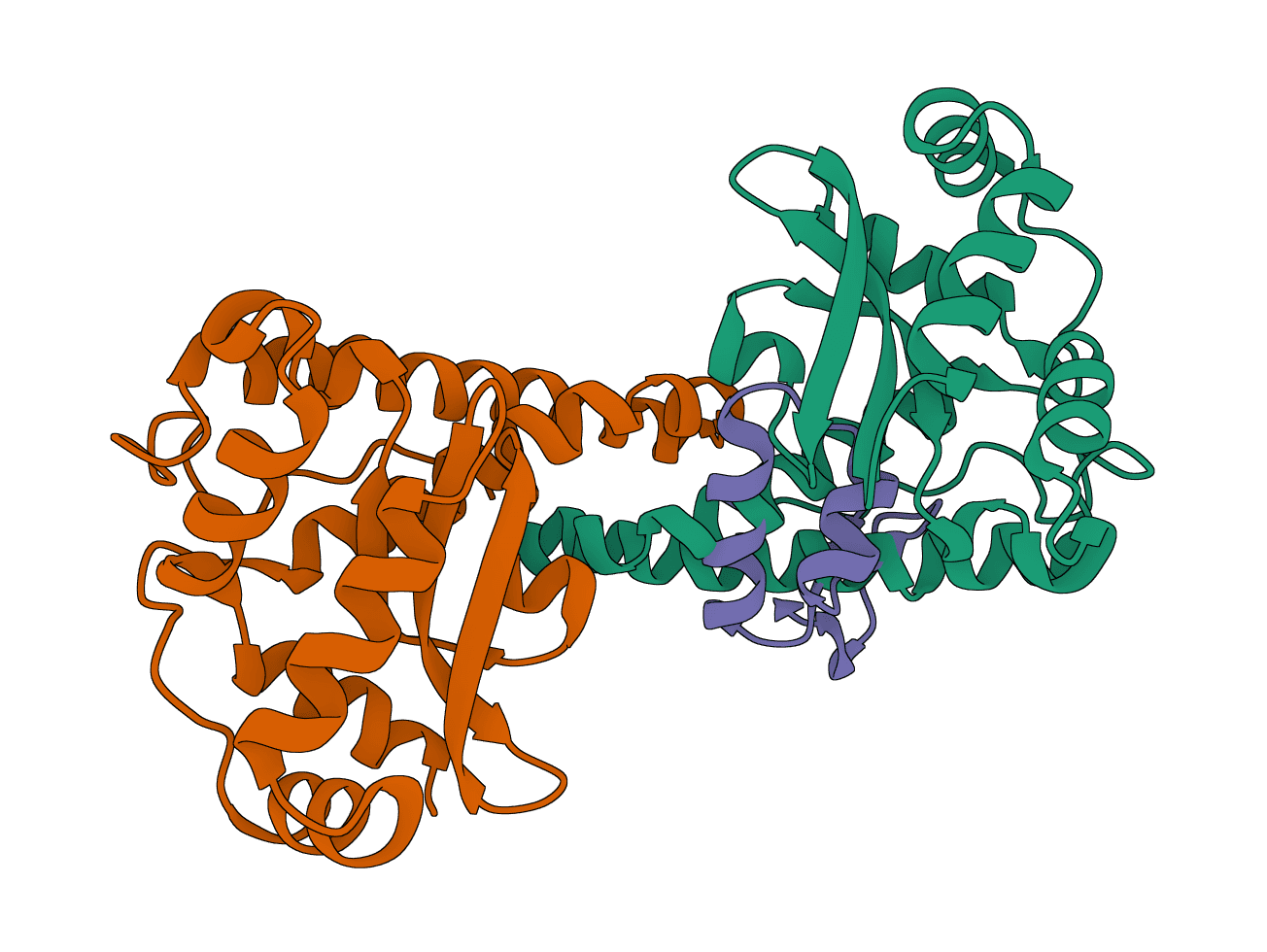
LightDock
LightDock is a protein-protein, protein-peptide, and protein-DNA docking framework using Glowworm Swarm Optimization (GSO). It predicts macromolecular binding modes and interfaces for biological complexes.
PandaDock
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
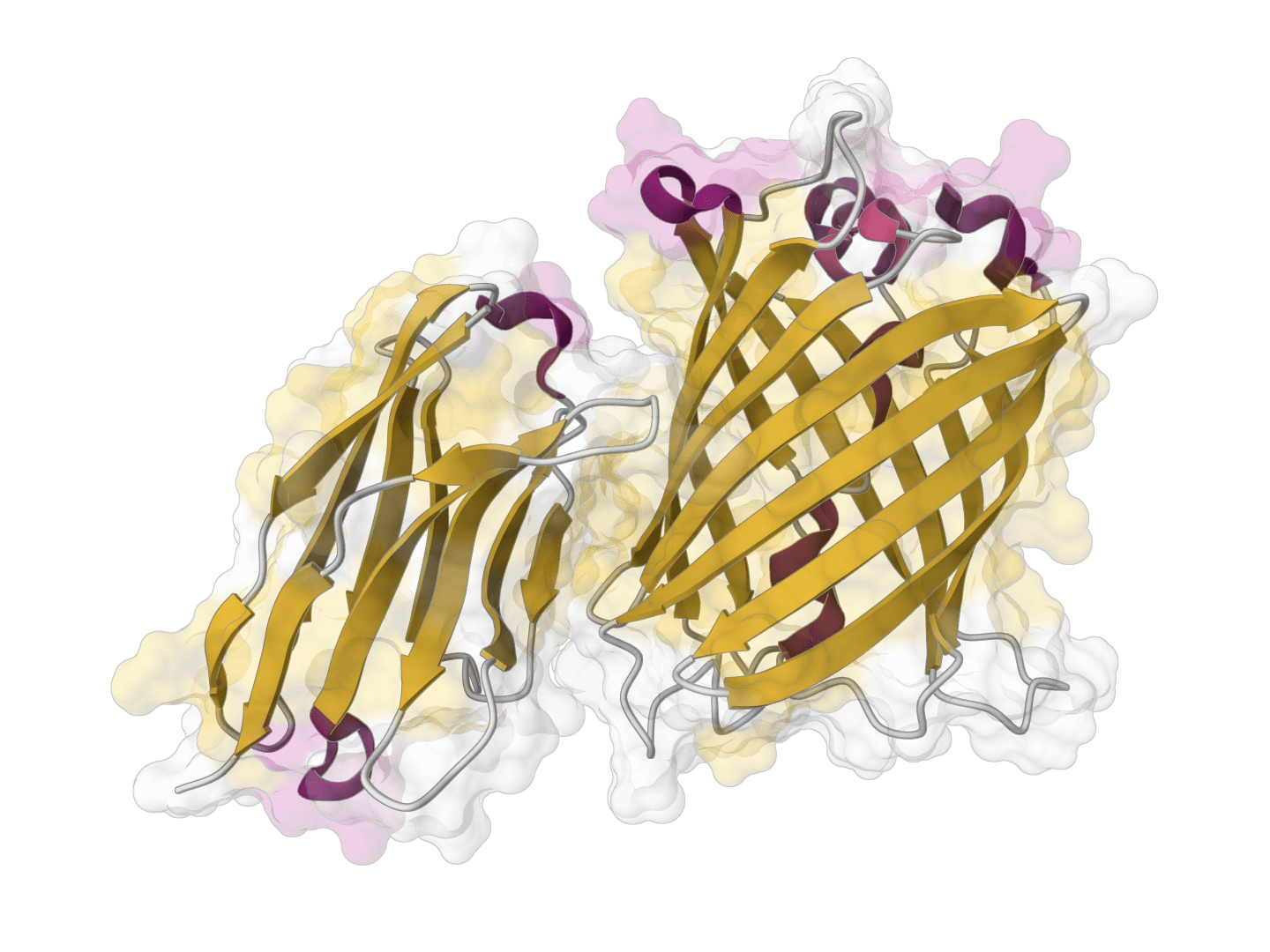
ParaSurf
ParaSurf is a state-of-the-art surface-based deep learning model for predicting interactions between antibodies and antigens. It identifies paratope binding sites on antibody structures with high accuracy across multiple benchmark datasets.
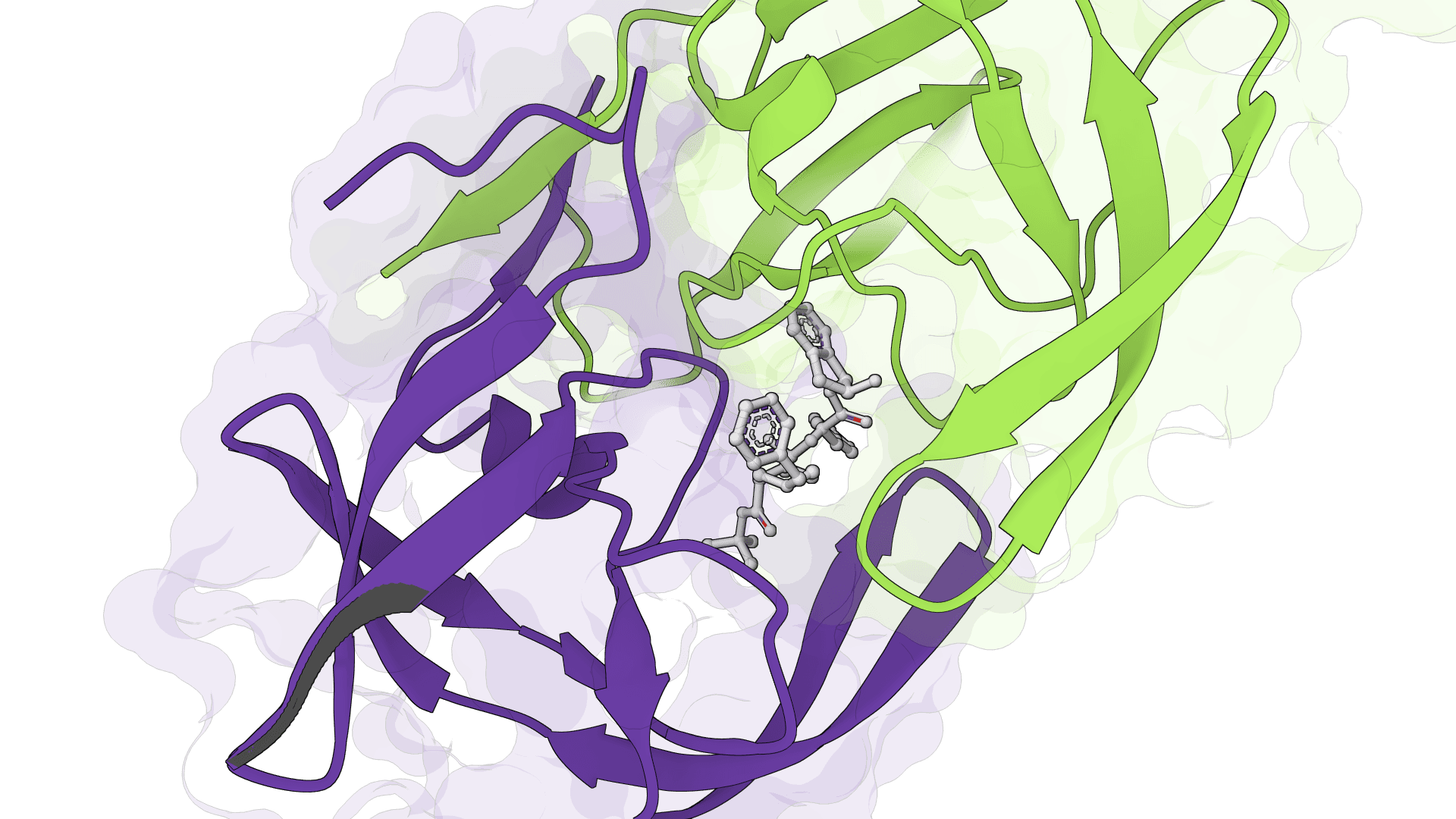
SigmaDock
SigmaDock is a fragment-based molecular docking tool using SE(3) equivariant diffusion models to predict how small molecule ligands bind to protein targets. Presented at ICLR 2026, it generates multiple binding poses with Vinardo scoring.
SMINA
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.
SurfDock
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.