Dock molecules with confidence
Screen protein-ligand and protein-protein interactions using AutoDock Vina, DiffDock, GNINA, and LightDock through a unified cloud platform.
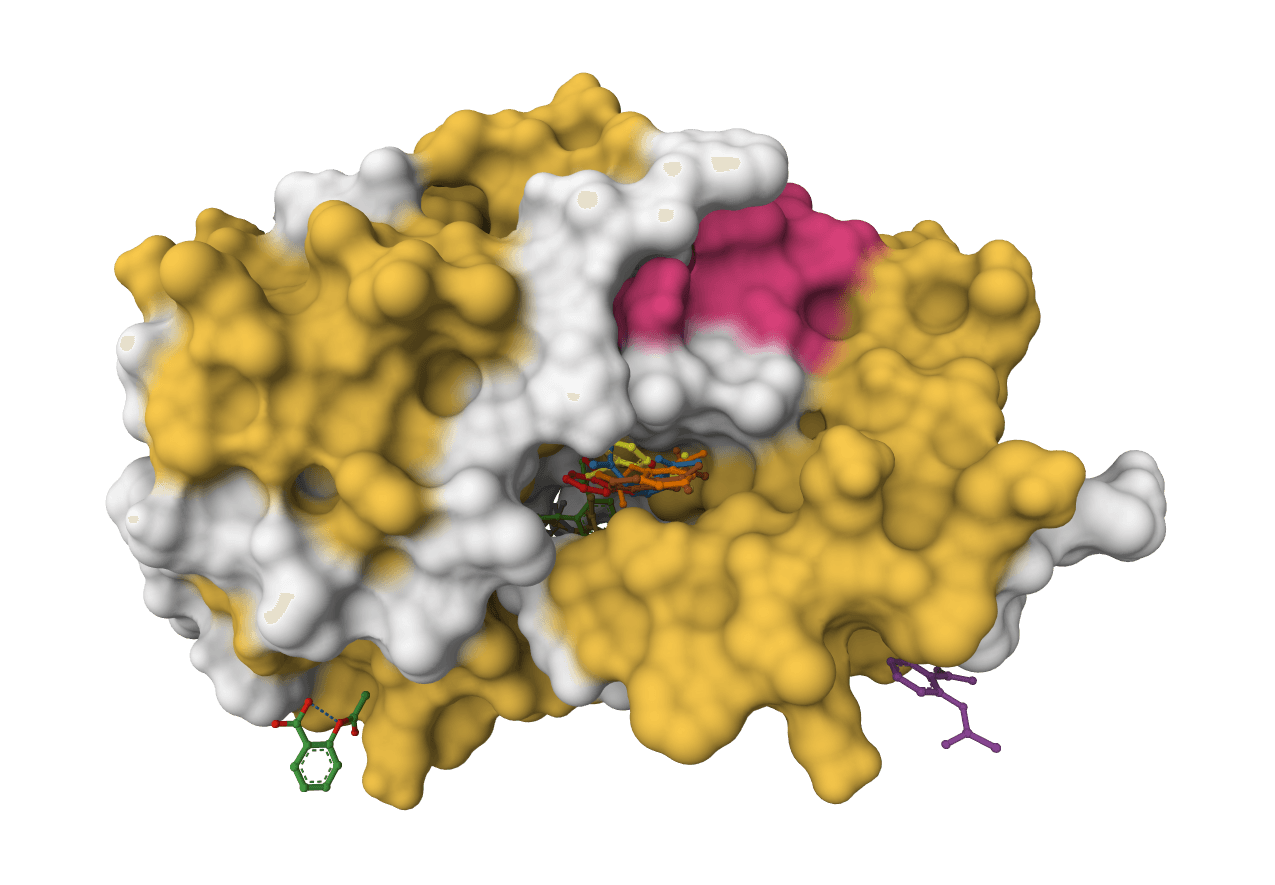
DiffDock-L
State-of-the-art molecular docking using diffusion models to predict how small molecule ligands bind to protein targets with confidence scores.
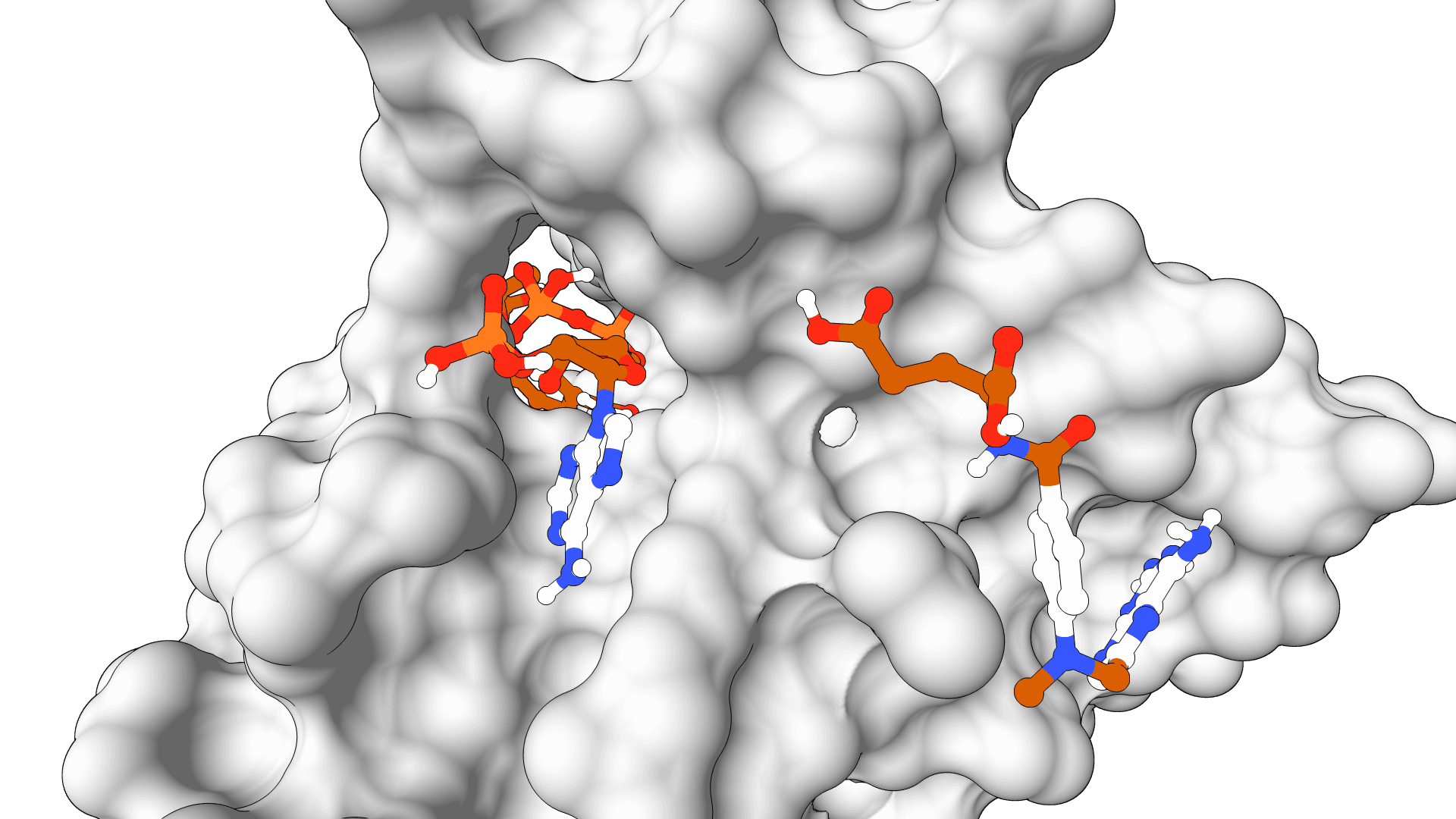
AutoDock-GPU
GPU-accelerated AutoDock for ultra-fast molecular docking. Screen thousands of compounds in minutes with CUDA acceleration.
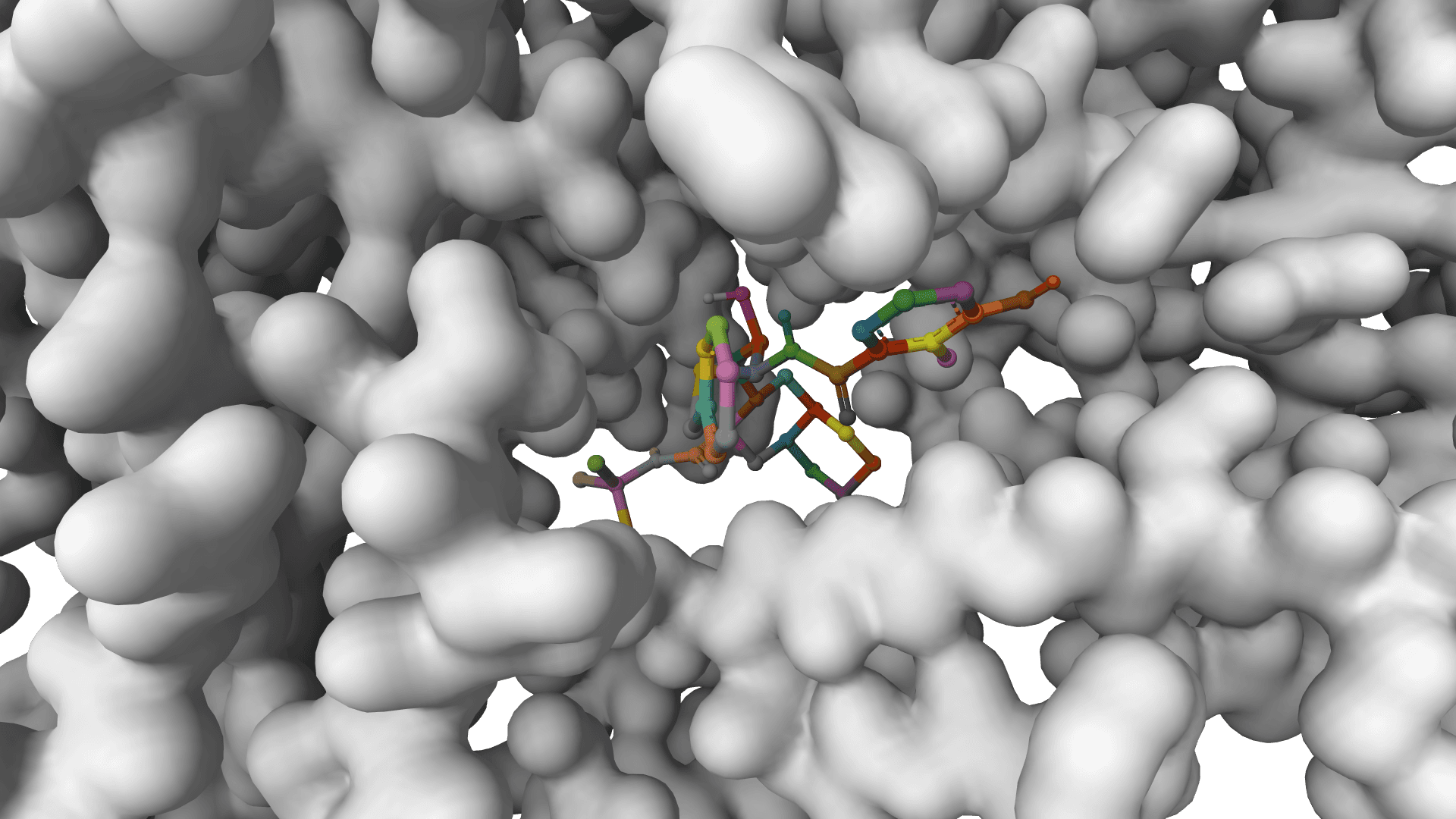
GNINA
Molecular docking combining physics-based scoring with deep learning CNN for accurate binding predictions with confidence.
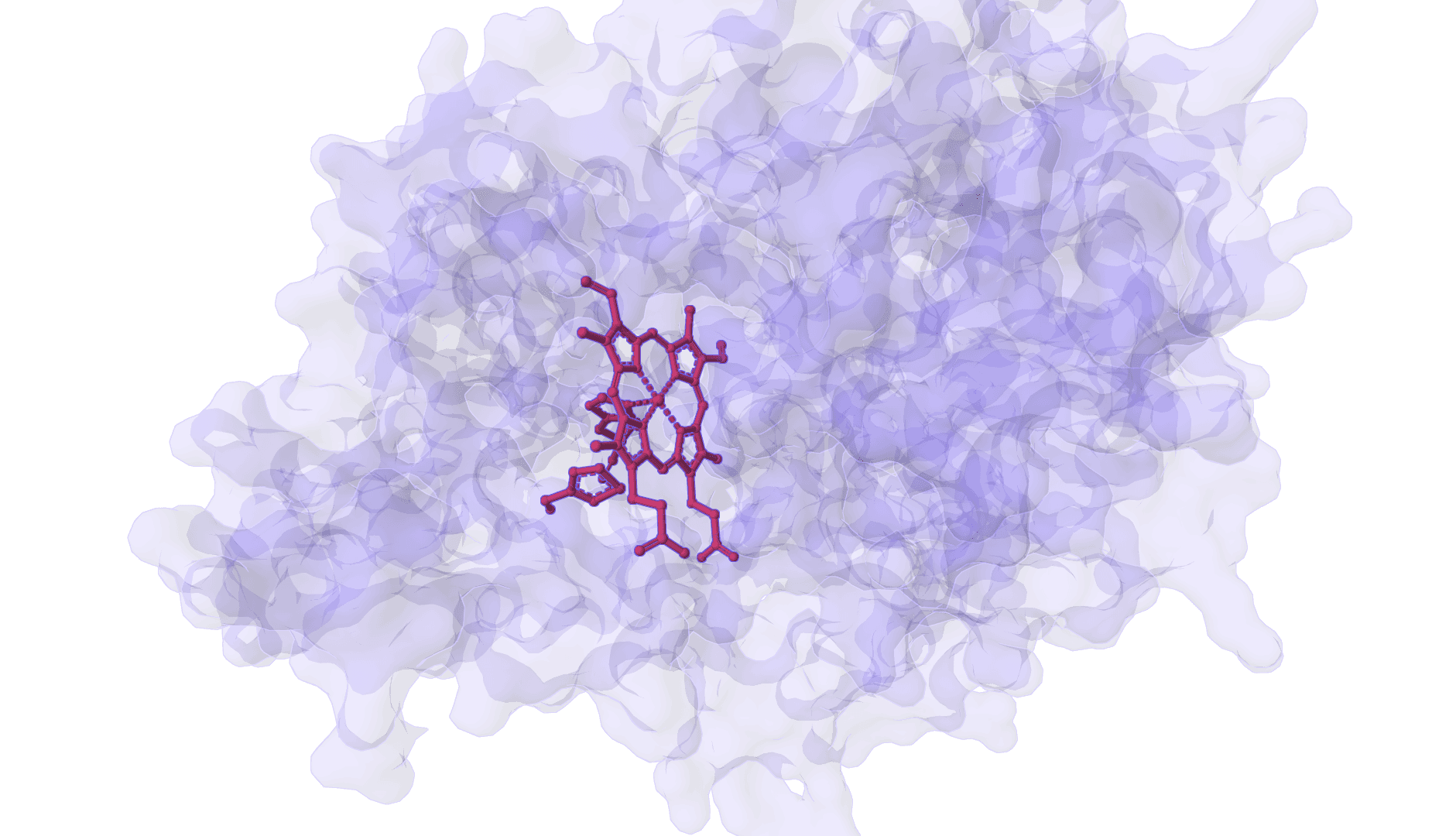
DynamicBind
AI-powered docking that predicts both ligand binding poses and protein conformational changes upon binding.
HADDOCK3
Information-driven protein-protein and protein-ligand docking. Integrates experimental data and AI predictions for accurate complex modeling.
LightDock
Protein-protein, protein-peptide, and protein-DNA docking using swarm optimization for macromolecular complex prediction.
Why researchers choose ProteinIQ
DiffDock, GNINA, and Vina run directly in your browser—zero setup
Traditional docking software requires complex installation and configuration
AI-powered blind docking finds binding sites automatically
Defining binding sites and grid boxes manually is error-prone
Parallel cloud computing docks thousands of compounds in hours
Processing large compound libraries takes weeks on local machines
Run multiple docking engines and compare results in one interface
Comparing results across different docking tools is tedious
How it works
High-performance molecular docking simulations using industry-standard tools like AutoDock Vina, DiffDock, GNINA, and LightDock. Predict protein-ligand and macromolecular binding affinity and poses.
Upload structures
Upload protein (PDB) and ligand (SDF/MOL2) files
Configure docking
Define binding site or use blind docking mode
Run docking
DiffDock or GNINA predicts binding poses and scores
Analyze results
Visualize poses, compare scores, export top hits
Related solutions
Discover complementary workflows to enhance your research.
Predict protein structures in minutes
Access AlphaFold3, Boltz, and Chai-1 models instantly—no infrastructure required, results in minutes. Design and refine structures with RFdiffusion, BoltzGen, and ProteinMPNN.
From molecules to medicines faster
Design and validate drug candidates with integrated ADMET-AI predictions, docking simulations, and lead optimization workflows.
Screen millions, discover one
Filter compound libraries at scale with QEPPI, EvoEF2, and PocketFlow to identify promising drug candidates efficiently.