Free
$0/mo
For trying ProteinIQ
Per user/month, billed annually
- 100 one-time credits
- Daily lightweight analysis
- Core sequence and format tools
Small molecule workflows
ProteinIQ helps you prepare targets, screen compound libraries, dock candidates, triage ADMET risk, and export the structures, scores, and files behind every decision.
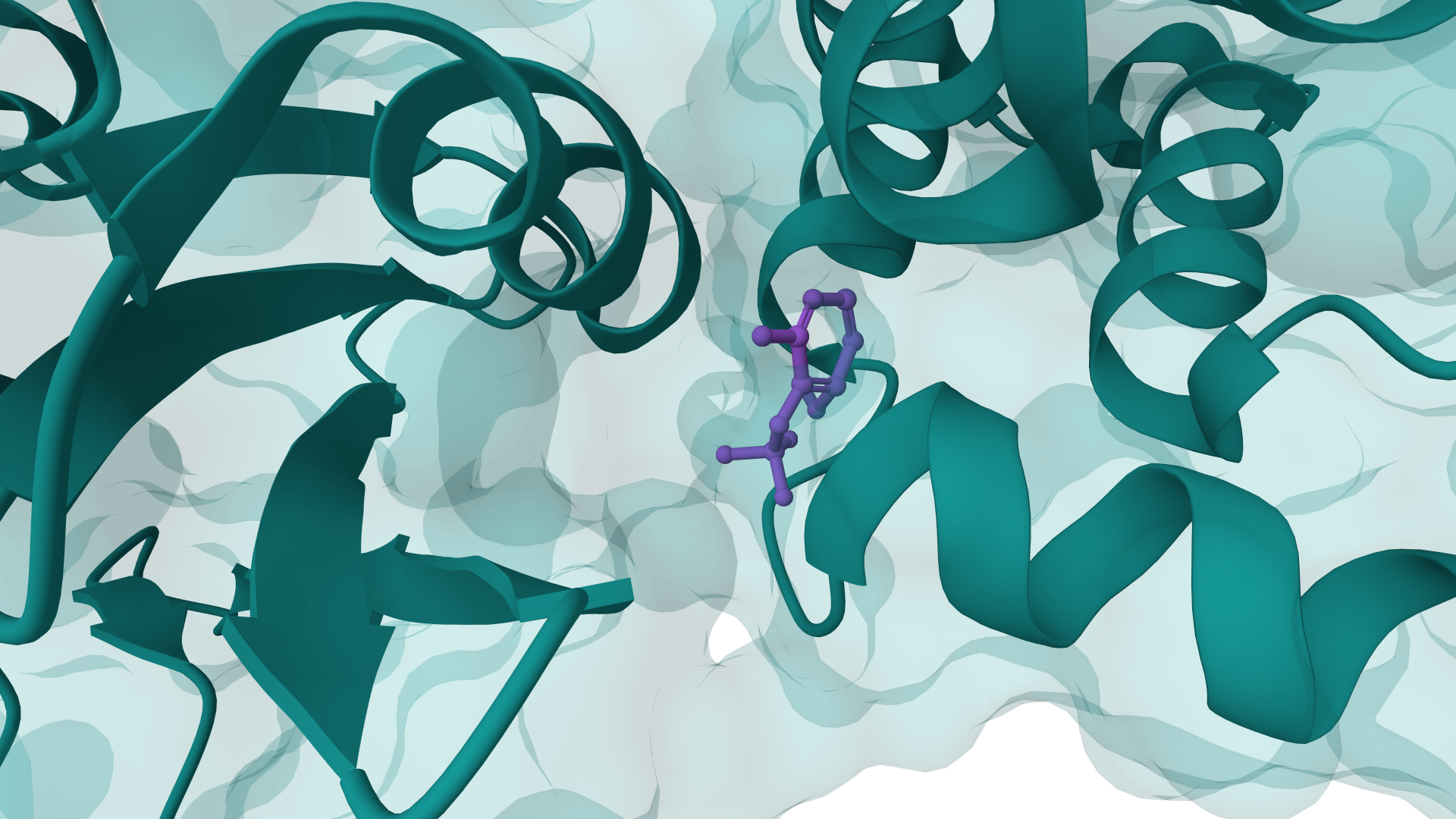
EGFR kinase with gefitinib
Interactive result · GNINA
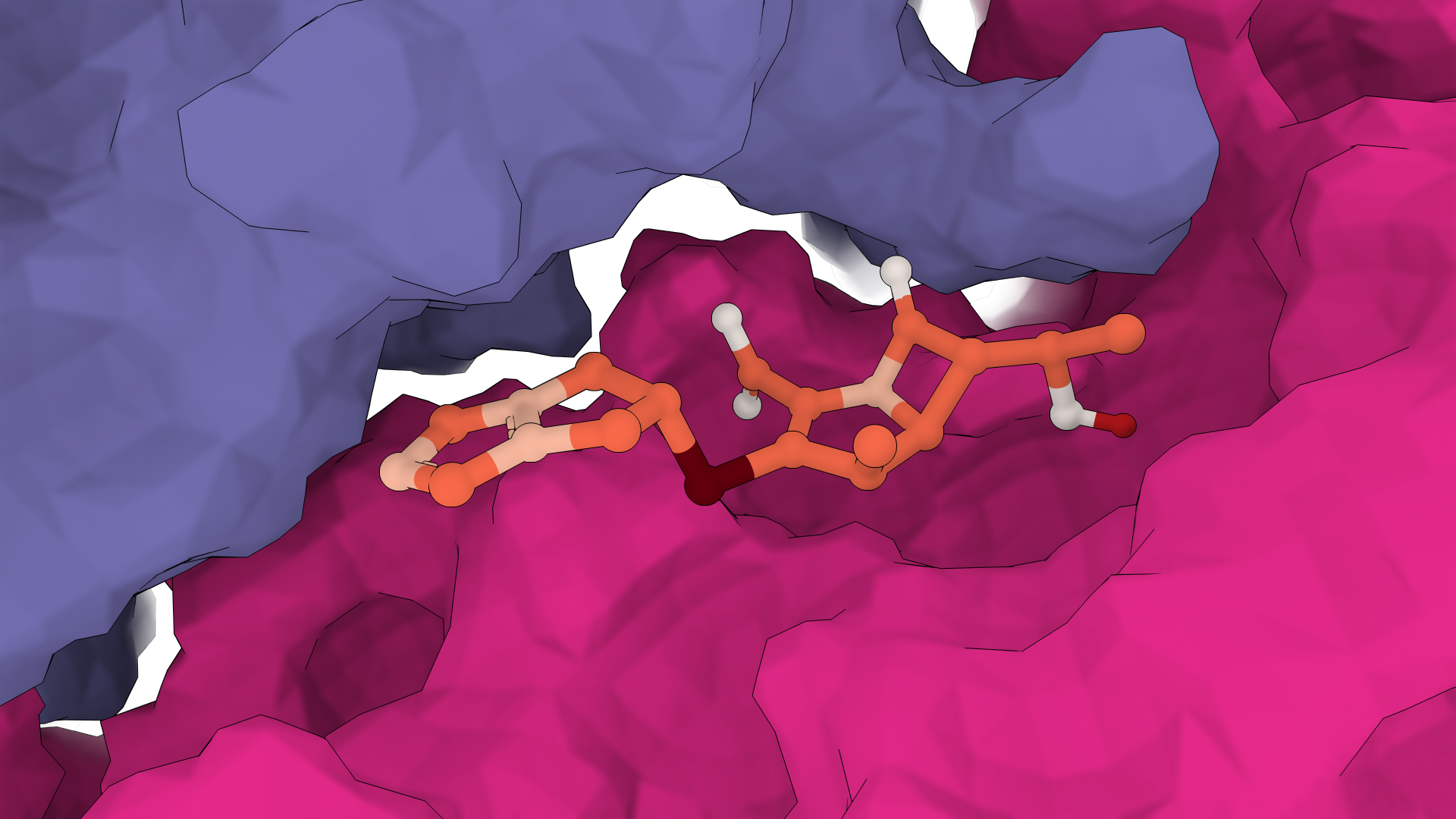
GNINA is a molecular docking tool that combines traditional physics-based docking with deep learning CNN scoring for protein-small-molecule complexes. It provides accurate binding predictions with confidence scores, optimized for high-throughput virtual screening.
Predict ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) properties from SMILES strings using machine learning models trained on Therapeutics Data Commons datasets.
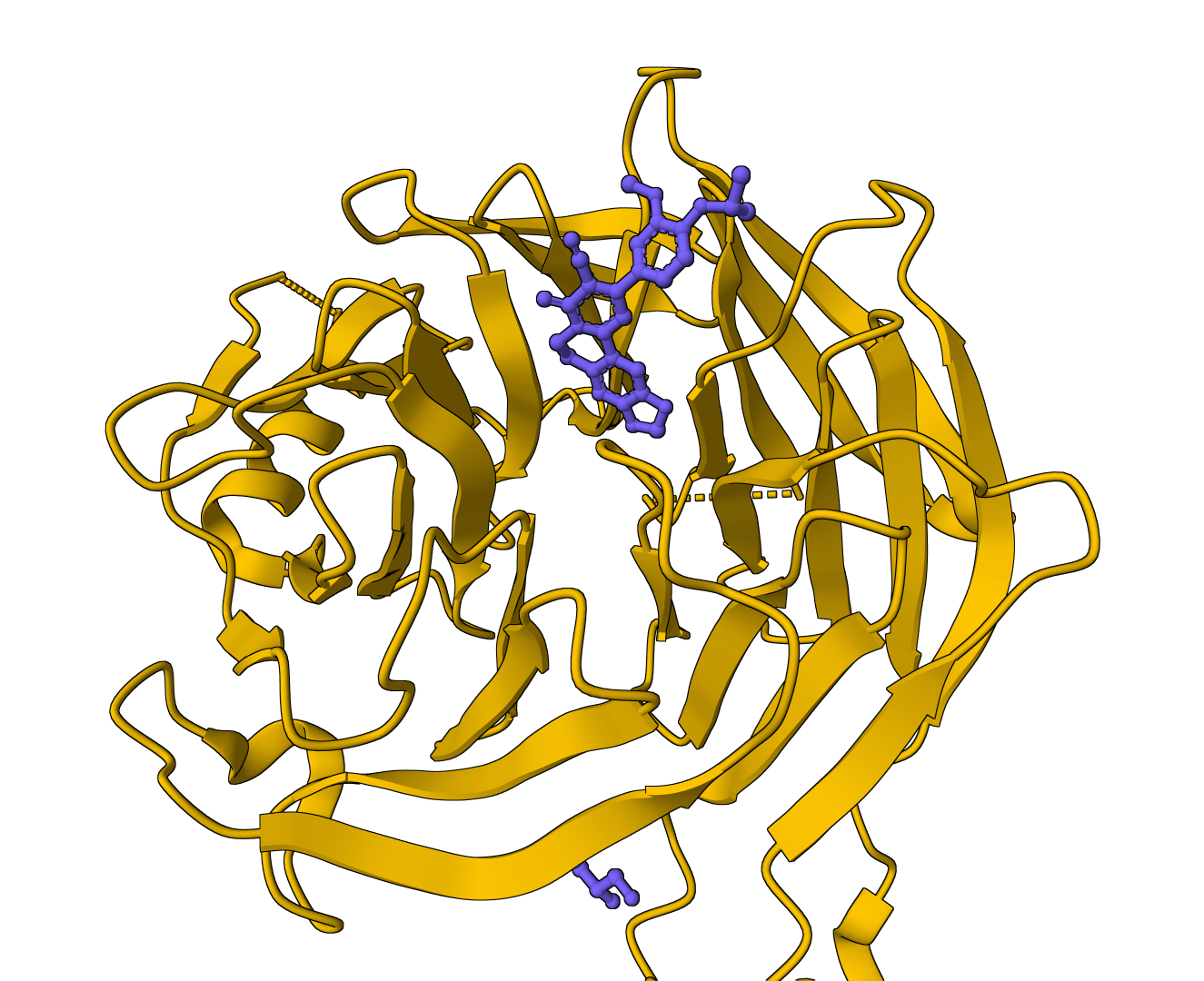
Open-source protein pocket detection using Voronoi tessellation and alpha spheres. Identifies ligand binding sites with druggability scores.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
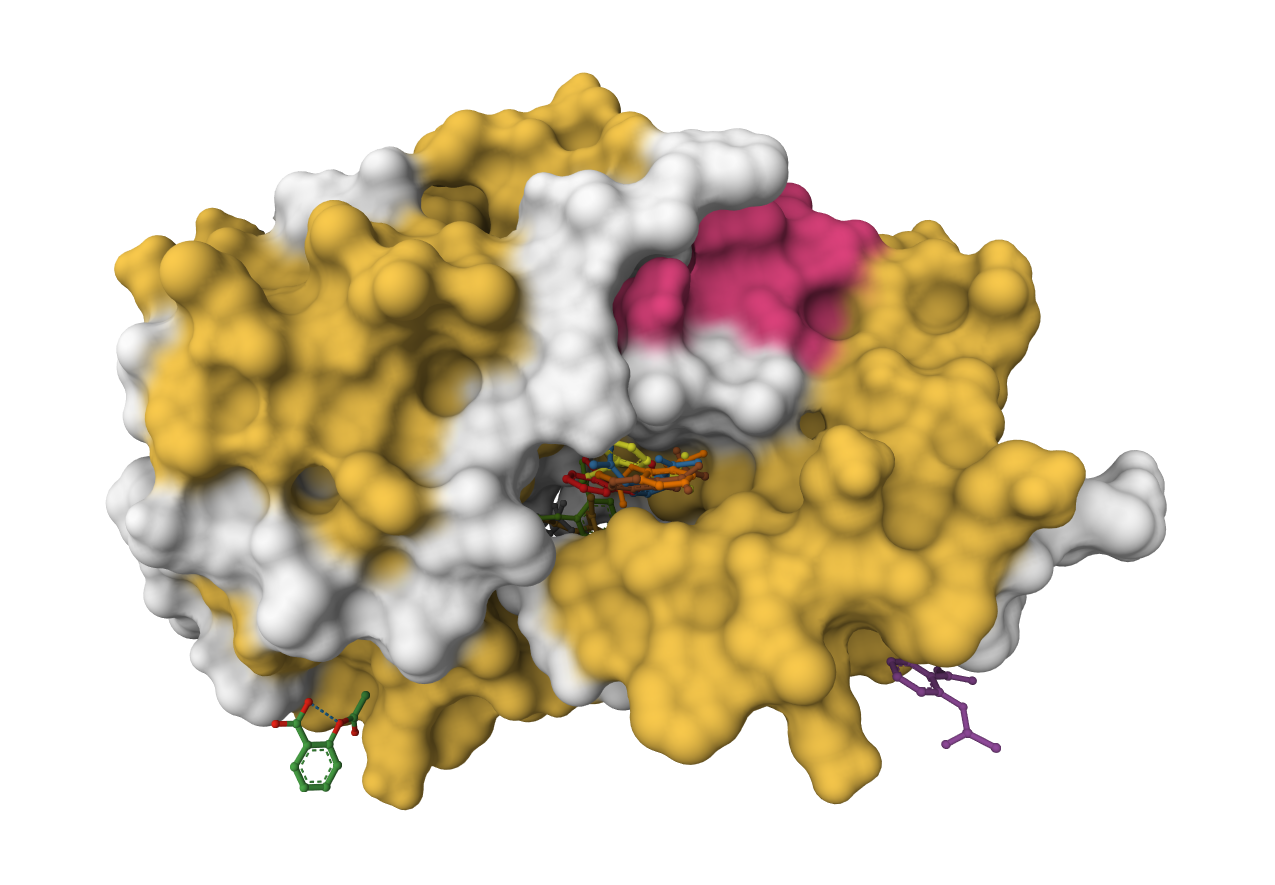
PocketFlow is a structure-based molecular generative model that designs novel drug-like molecules within protein binding pockets. It uses autoregressive flow modeling with chemical knowledge to generate 100% chemically valid, highly drug-like compounds.
PoseBusters validates generated or docked molecular poses with chemically and structurally grounded quality checks for molecular geometry, intermolecular interactions, and optional reference-pose agreement.
For trying ProteinIQ
Per user/month, billed annually
For academics
Per user/month, billed annually
For commercial research
Per user/month, billed annually
For organizations needing custom control
Custom annual terms
ProteinIQ supports small molecule drug discovery workflows for target preparation, ligand preparation, property filtering, docking, ADMET review, toxicity screening, and follow-up structure analysis. You can run the steps as a connected workflow or open the individual upstream tools when you only need one analysis.
ProteinIQ virtual screening tools commonly start from a target structure or model plus ligands in formats such as SMILES, SDF, MOL2, PDB, or tabular compound files. The exact input requirements stay tied to the upstream tool, so docking, filtering, and property prediction steps preserve their native constraints.
Yes. ProteinIQ can help prepare targets and ligands, run filters, dock selected compounds, compare ranked poses, and export a shortlist for review. The platform is designed for computational virtual screening and prioritization, not for silently changing upstream docking scores or replacing experimental follow-up.
ADMET and toxicity prediction in ProteinIQ can be used before docking to reduce a large compound library, after docking to compare top-ranked molecules, or both. These outputs are computational prioritization signals, so ProteinIQ keeps the prediction tables inspectable rather than treating them as experimental validation.
ProteinIQ displays molecular docking results as the upstream tool produces them, including ranked poses, score tables, structure files, logs, and downloadable artifacts where available. The goal is to make docking results easier to compare without changing scoring conventions or hiding the original output files.
Yes. ProteinIQ workflows are useful when you want connected target preparation, ligand filtering, docking, and triage steps, but individual small molecule tools remain available when you only need one analysis. This lets you use ProteinIQ as a workflow layer or as a code-free launcher for a specific upstream tool.
ProteinIQ exports the outputs produced by each small molecule tool, which can include prepared structures, ranked pose files, docking score tables, CSV screening results, logs, and upstream result files. Export support depends on the selected tool, but the platform is built to keep downloadable evidence attached to the compound or target it came from.
No. ProteinIQ helps organize computational screening, molecular docking, ADMET prediction, and toxicity prioritization before experimental follow-up. Experimental binding, ADMET, toxicity, selectivity, and developability assays are still required before making biological or development claims.
You can start in ProteinIQ by choosing a workflow template for docking or screening, or by opening a specific tool such as docking, pocket detection, ADMET prediction, or pose validation. The practical first step is to prepare a target structure and a small ligand set, then expand into larger screening runs after the inputs look correct.