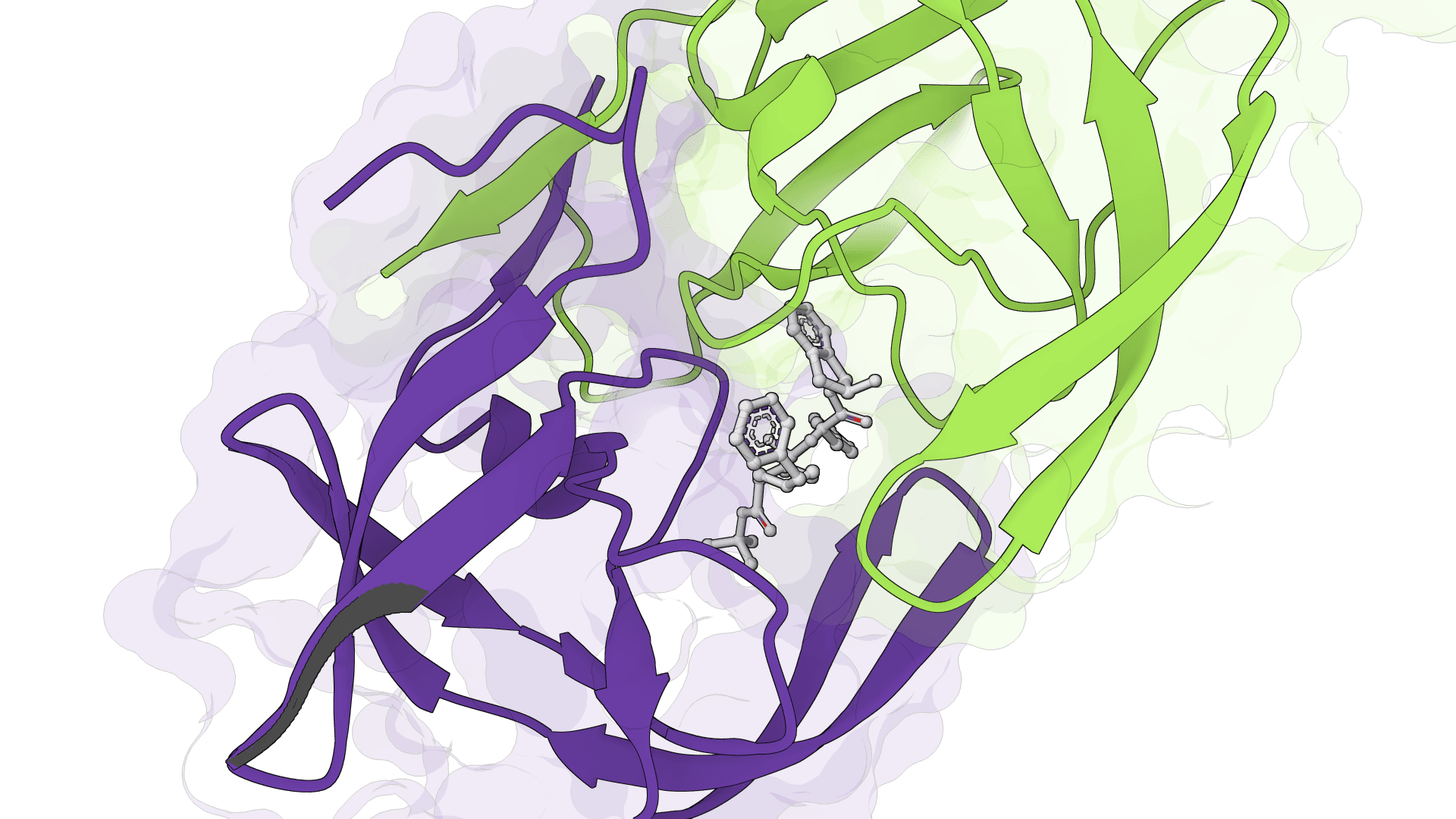
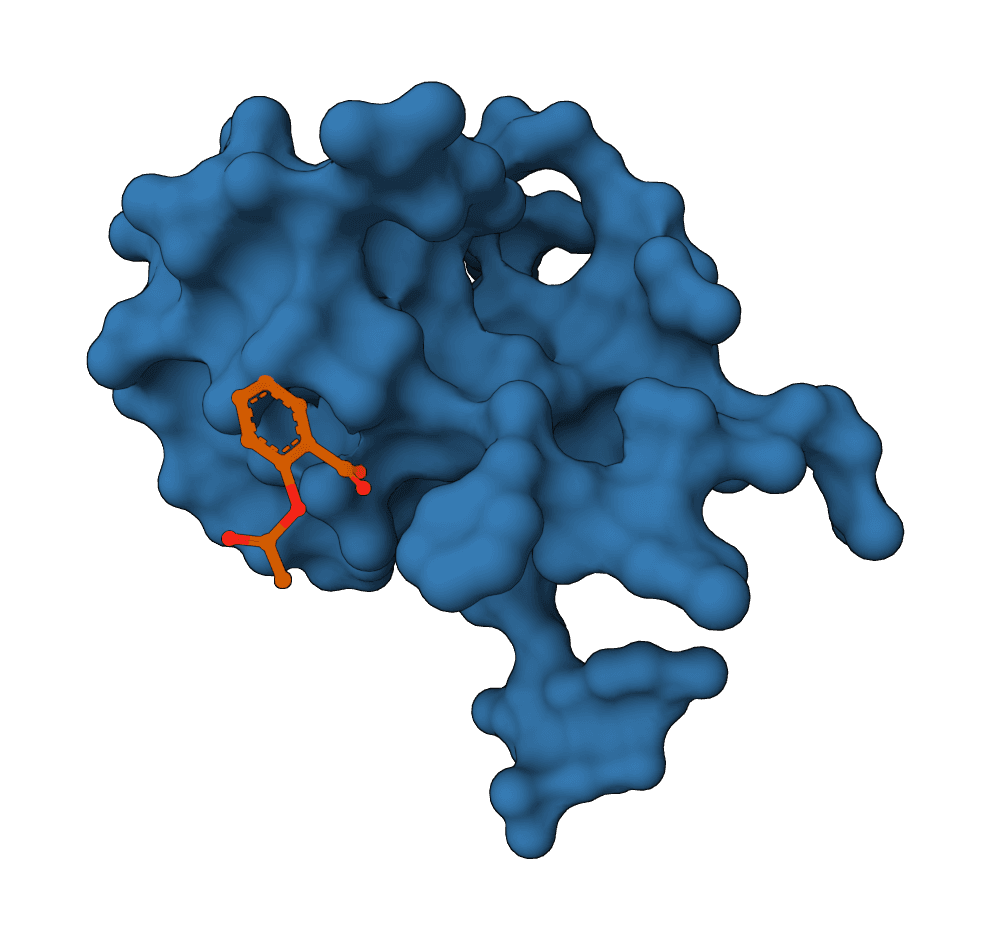
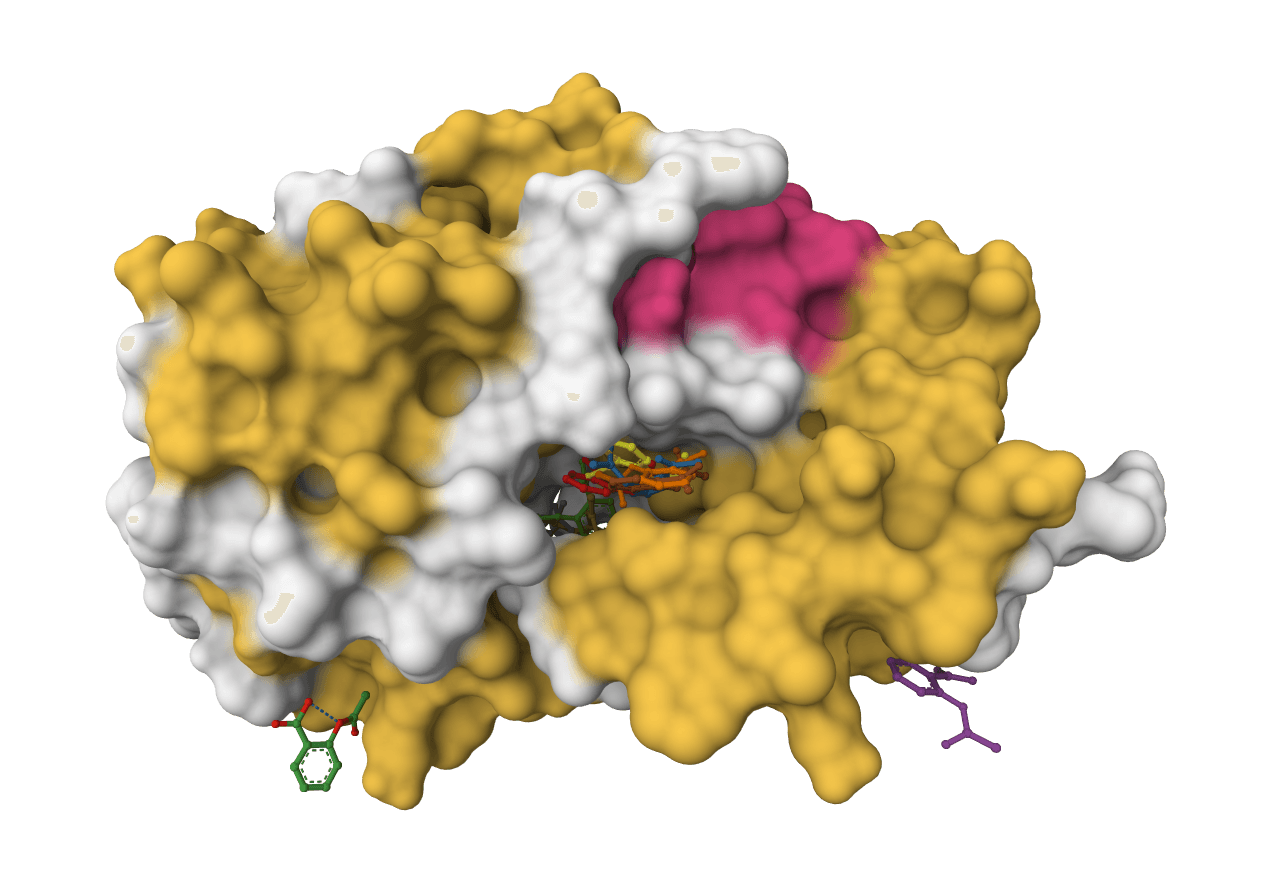
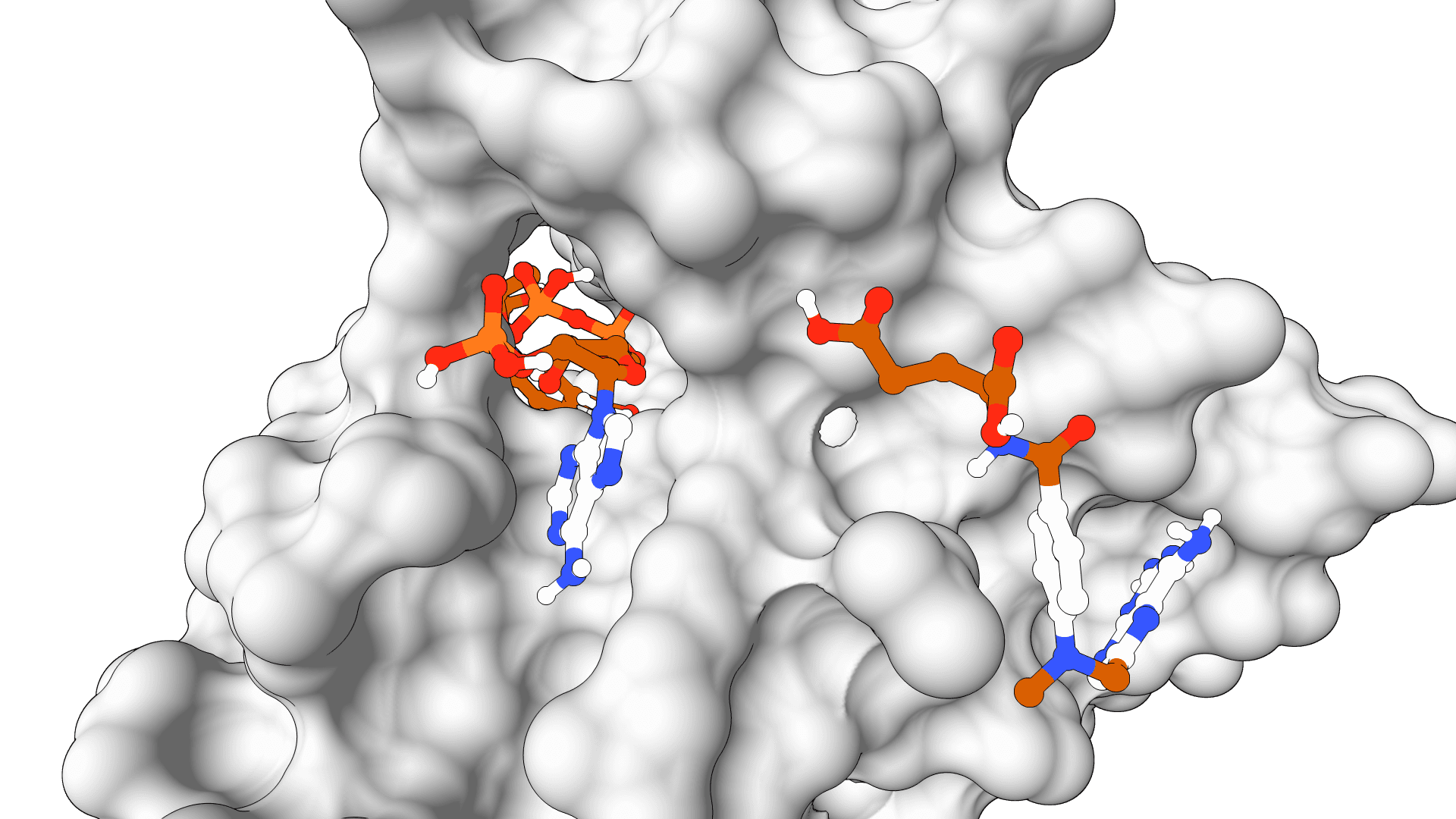
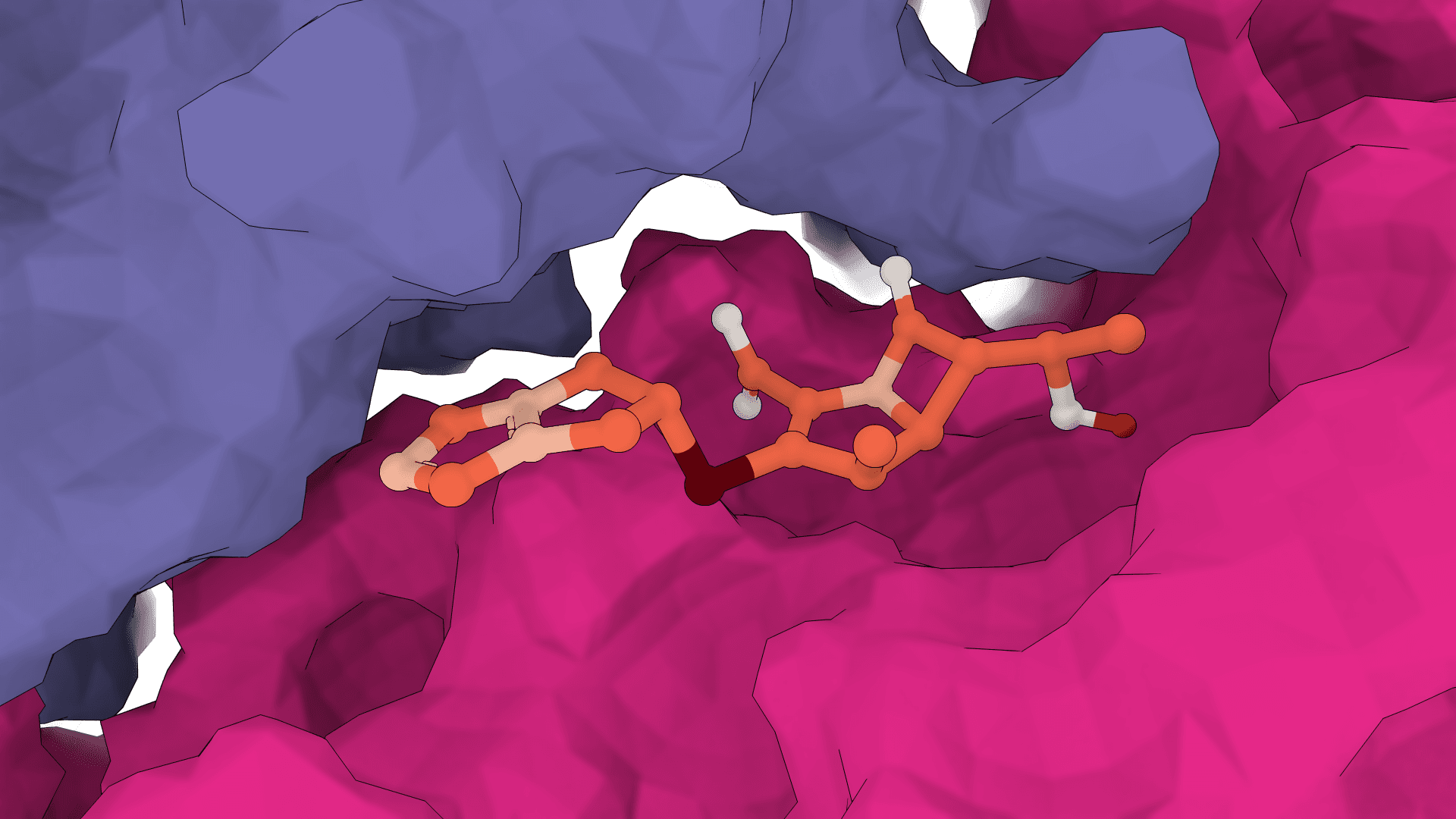
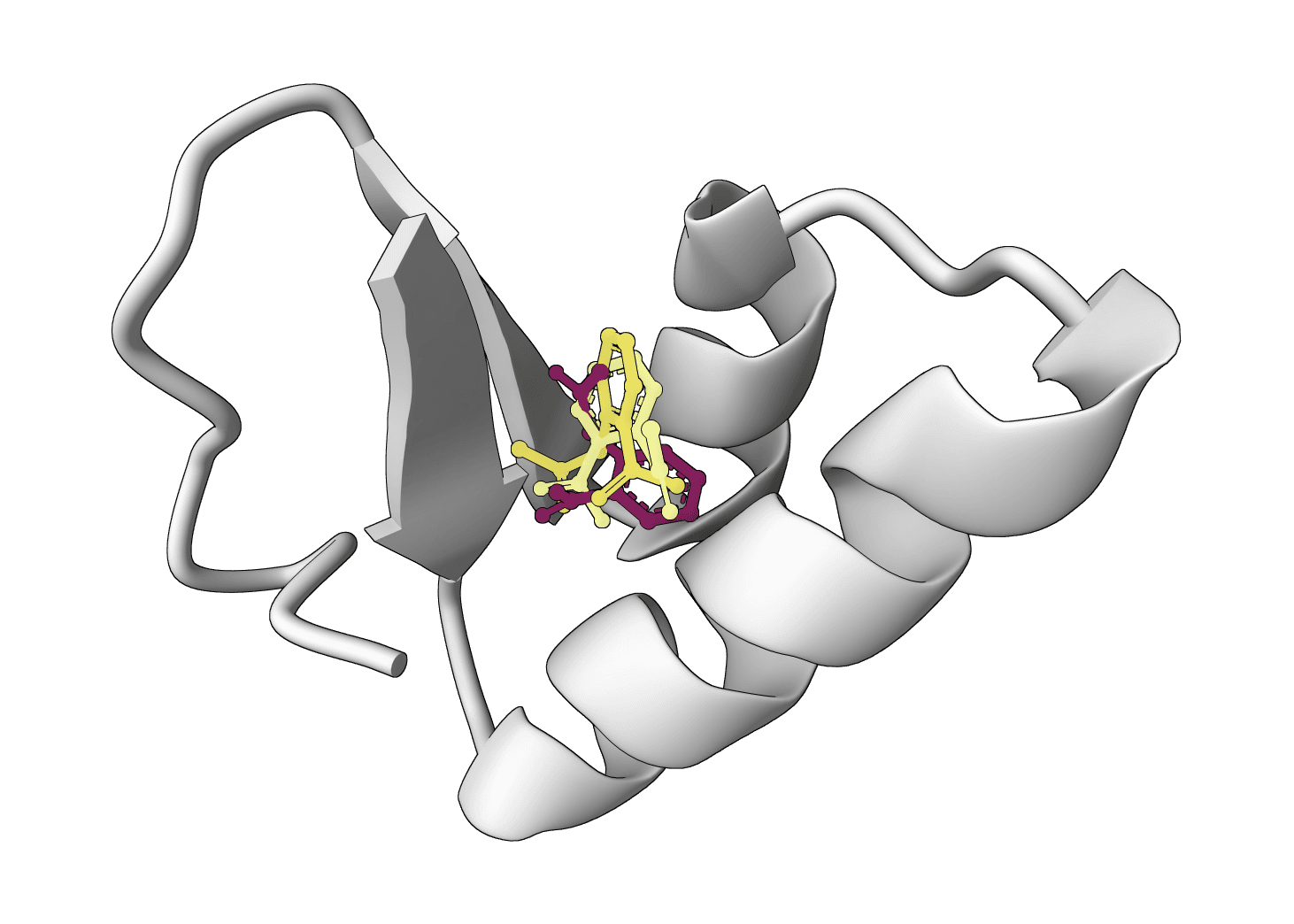What is SigmaDock?
SigmaDock is a molecular docking method that predicts how small molecule ligands bind to protein targets. Developed at the University of Oxford and presented at ICLR 2026, it achieves approximately 80% Top-1 success rate (RMSD <2 Å and PB-valid) on the PoseBusters benchmark — the first deep learning method to surpass classical physics-based docking on this metric.
Rather than treating ligand flexibility through torsional angles, SigmaDock decomposes ligands into rigid-body fragments at their rotatable bonds and learns SE(3) transformations for each fragment. This design improves both the physical validity of generated poses and the model's ability to generalize to proteins not seen during training.
How to use SigmaDock online
ProteinIQ hosts SigmaDock on GPU infrastructure, so no software installation or local GPU is needed. Submit a protein structure and a ligand, and the model generates multiple ranked binding poses within minutes.
SigmaDock expects a standard protein receptor structure that native parsing tools can read cleanly. Receptors with malformed coordinates, unusual fibril assemblies, or heavily modified/non-standard residues may be rejected before docking rather than producing unreliable poses.
Settings
Docking parameters
Binding site
SigmaDock requires a known binding pocket — it is not a blind docking method. By default, the binding site is automatically determined from the co-crystallized ligand in the PDB file. For structures without a bound ligand (e.g., AlphaFold predictions or apo crystal structures), the pocket center can be specified manually.
Results
Each run produces a set of ranked poses as SDF files, displayed in a 3D interactive viewer alongside the input protein. A spreadsheet summarizes the key metrics for each pose.
Interpreting scores
The primary Score column reflects the chosen scoring function (Vinardo or Vina). As a general guide for Vinardo/Vina scores:
The CNN Score provides an orthogonal assessment: poses where both the physics-based score and the CNN score agree tend to be more reliable. A CNN Score above 0.5 combined with a strong Vinardo affinity is a good indicator. The PB Valid score flags poses that may look energetically favorable but contain geometric distortions — a PB Valid score below 0.7 warrants visual inspection of the pose.
How SigmaDock works
Molecular docking requires sampling a very large conformational space efficiently. Classical methods like Vina grid-search this space using empirical force fields; diffusion models like DiffDock operate in torsional angle space. SigmaDock takes a different route.
Fragment decomposition
A ligand is broken at its rotatable bonds into rigid fragments. Each fragment maintains its fixed internal geometry (bond lengths, angles) throughout the docking process. The docking problem is then reframed as predicting an SE(3) transformation (translation + rotation in 3D space) for each fragment rather than a set of torsional angles.
SE(3) diffusion
The model uses Riemannian diffusion on the SE(3) manifold. During training, fragment poses are progressively randomized via Brownian motion on the translation and rotation spaces. The neural network learns to reverse this process — denoising noisy fragment configurations back into plausible bound poses. At inference, docking starts from randomized fragment poses and iteratively applies the learned denoising steps.
Architecture
SigmaDock extends EquiformerV2, a graph transformer with SO(3)-equivariance, with a hierarchical graph topology using virtual nodes and edges to capture multi-scale protein-ligand interactions. The prediction head is SO(3)-equivariant to avoid local coordinate frame ambiguities. Soft geometric constraints via triangulation distance conditioning enforce chemical validity across fragment junctions throughout the diffusion trajectory.
Inductive biases over scale
SigmaDock intentionally uses a relatively small training set (~19,000 examples) but achieves AlphaFold3-comparable accuracy through well-designed inductive biases: equivariance, fragment rigidity, and geometric constraints. This contrasts with purely data-driven approaches that rely on much larger training corpora.
On the PoseBusters benchmark with a temporal train/test split:
SigmaDock also maintains strong performance on proteins with low sequence similarity to its training set, a common failure mode for deep learning docking methods.
Limitations
- Not a blind docking method — a binding site must be known. The tool auto-detects it from co-crystallized ligands in the PDB, or coordinates can be supplied manually.
- Protein backbone is held rigid; conformational changes upon ligand binding (induced fit) are not modeled.
- Receptor parsing is strict. Standard protein PDBs work best; malformed exports, fibrils, and structures with modified/non-standard residues may need cleanup before submission.
- Metal ions and metal-coordinating ligands are not supported; the method is designed for organic small molecules.
- Proteins over 1000 residues must be trimmed to the binding domain before submission.