IntelliFold 2 is an all-atom biomolecular structure prediction model for proteins, ligands, DNA, RNA, and mixed complexes. It extends the earlier IntFold foundation model with an updated architecture and a faster v2-Flash inference path, while retaining the project's focus on controllable prediction for practical design and screening workflows.
The IntFold technical report describes the platform as a controllable foundation model that reaches AlphaFold 3 level performance on broad biomolecular benchmarks while adding mechanisms for structural constraints, allosteric-state guidance, and downstream ranking. The IntelliFold 2 release positions the newer model family as an architectural refinement intended to improve accuracy on recent FoldBench evaluations.
ProteinIQ provides browser-based access to IntelliFold 2, so the model can be run without local GPU setup, Python installation, or manual environment configuration. Jobs are submitted from a single form that combines molecular inputs, MSA options, and inference settings.
At least one foldable biopolymer is required in practice: a protein, DNA, or RNA chain. The ProteinIQ deployment accepts up to 10 entries for each molecule type, with a combined maximum of 3,000 residues across all protein, DNA, and RNA chains.
| Input | Description |
|---|---|
Protein | FASTA text, .fasta/.fa upload, or an RCSB sequence fetch. Up to 10 chains. |
Precomputed MSA | Optional .a3m or paired .csv alignment files. Up to 10 files. |
Ligand (SMILES) | SMILES entered directly or fetched from PubChem. Up to 10 ligands. |
Ligand (CCD) | PDB Chemical Component Dictionary codes such as ATP, NAD, or SAH. Up to 10 ligands. |
DNA | FASTA text or .fasta/.fa upload. Up to 10 chains. |
RNA | FASTA text or .fasta/.fa upload. Up to 10 chains. |
Job name | Optional label used to identify the run in job history. |
| Setting | Description |
|---|---|
Model | Chooses v2-Flash (default), v2, or v1. v2-Flash is the recommended speed and quality trade-off. |
Diffusion samples | Structures generated per seed (1-10, default 5). Higher values broaden conformational sampling but increase runtime. |
Recycling iterations | Iterative refinement cycles during inference (1-12, default 10). More recycling can improve difficult predictions. |
Sampling steps | Diffusion denoising steps (50-300, default 200). Higher values may improve structure quality at the cost of latency. |
Seeds | One seed or a comma-separated list such as 42,43,44. Multiple seeds produce larger ranked ensembles. |
Output format | mmCIF (default) or PDB. mmCIF is generally preferable for modern structure workflows. |
| Setting | Description |
|---|---|
Generate MSA with server | Uses the MMseqs2 server to build protein MSAs automatically. Disable for single-sequence runs or when uploading precomputed alignments. |
MSA pairing strategy | Pairing mode for multimer alignments when server-side MSA generation is enabled: Greedy (default) or Complete. |
Disable pairing | Turns off paired MSA generation for multimer jobs when only unpaired alignments are desired. |
ProteinIQ returns an interactive structure viewer, a downloadable results table, and the full file bundle for each run.
| Output | Description |
|---|---|
Viewer | Interactive 3D visualization of predicted structures. |
Data table | Ranked prediction summary derived from the primary output files. |
Structure files | Predicted models in mmCIF or PDB, depending on the selected output format. |
Download bundle | All result files produced by the run, including ranked predictions and associated metadata. |
The IntFold project is built as an all-atom foundation model for biomolecular structure prediction. In the authors' description, its distinguishing feature is controllability: the same core system is designed to support general structure prediction as well as guided tasks such as constrained inference, alternative-state modeling, and binding-focused applications.
The IntFold technical report describes specialized adapters that can bias the model toward user-defined structural constraints, allosteric states, and affinity-related downstream tasks. That makes IntelliFold different from sequence-to-structure systems that only return an unconstrained best guess.
ProteinIQ supports the model's diffusion and recycling controls directly. Diffusion sampling starts from noisy coordinates and refines them over many denoising steps, while recycling passes intermediate predictions back through the network for further correction. Increasing either parameter usually improves search depth, but the trade-off is longer runtime.
IntelliFold 2 can use server-generated MSAs through MMseqs2 or run from sequence input with MSA generation disabled. In protein systems with clear evolutionary homologs, MSA depth usually improves structural confidence; disabling MSA is more appropriate for speed-sensitive runs or synthetic targets with weak sequence family coverage.
The IntFold paper also describes a training-free, similarity-based ranking strategy for selecting better predictions from an ensemble. On ProteinIQ, that ensemble is controlled indirectly through Diffusion samples and Seeds, which determine how many candidate structures are generated before ranking.
IntelliFold 2 predictions are typically examined as a ranked set rather than a single definitive structure. Confidence metrics included in the IntelliFold 2 release materials and ProteinIQ configuration are the most useful first-pass filters.
| Metric | Interpretation |
|---|---|
pLDDT | Per-residue confidence on a 0-100 scale. Higher values indicate more reliable local geometry. |
pTM | Predicted template modeling score for overall fold quality. Higher values suggest a more reliable global structure. |
ipTM | Interface confidence for multichain complexes. Most useful when judging protein-protein or protein-nucleic acid assemblies. |
For exploratory jobs, multiple seeds and diffusion samples are often more informative than a single large run with fixed randomness. If top-ranked structures differ substantially from one another, the target may be flexible, weakly constrained by sequence evidence, or genuinely multimodal.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
OpenFold-3 is an open-source AI model for biomolecular structure prediction, aiming to reproduce AlphaFold3. Predicts 3D structures for proteins, RNA, DNA, and small molecule ligands with high accuracy.
Enhanced Protenix v2 biomolecular structure prediction by ByteDance. Predicts 3D structures for proteins, RNA, DNA, and small molecule ligands with high accuracy.
Open-source structure prediction neural network for proteins, nucleic acids, and small molecules. State-of-the-art accuracy with multi-chain support.
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
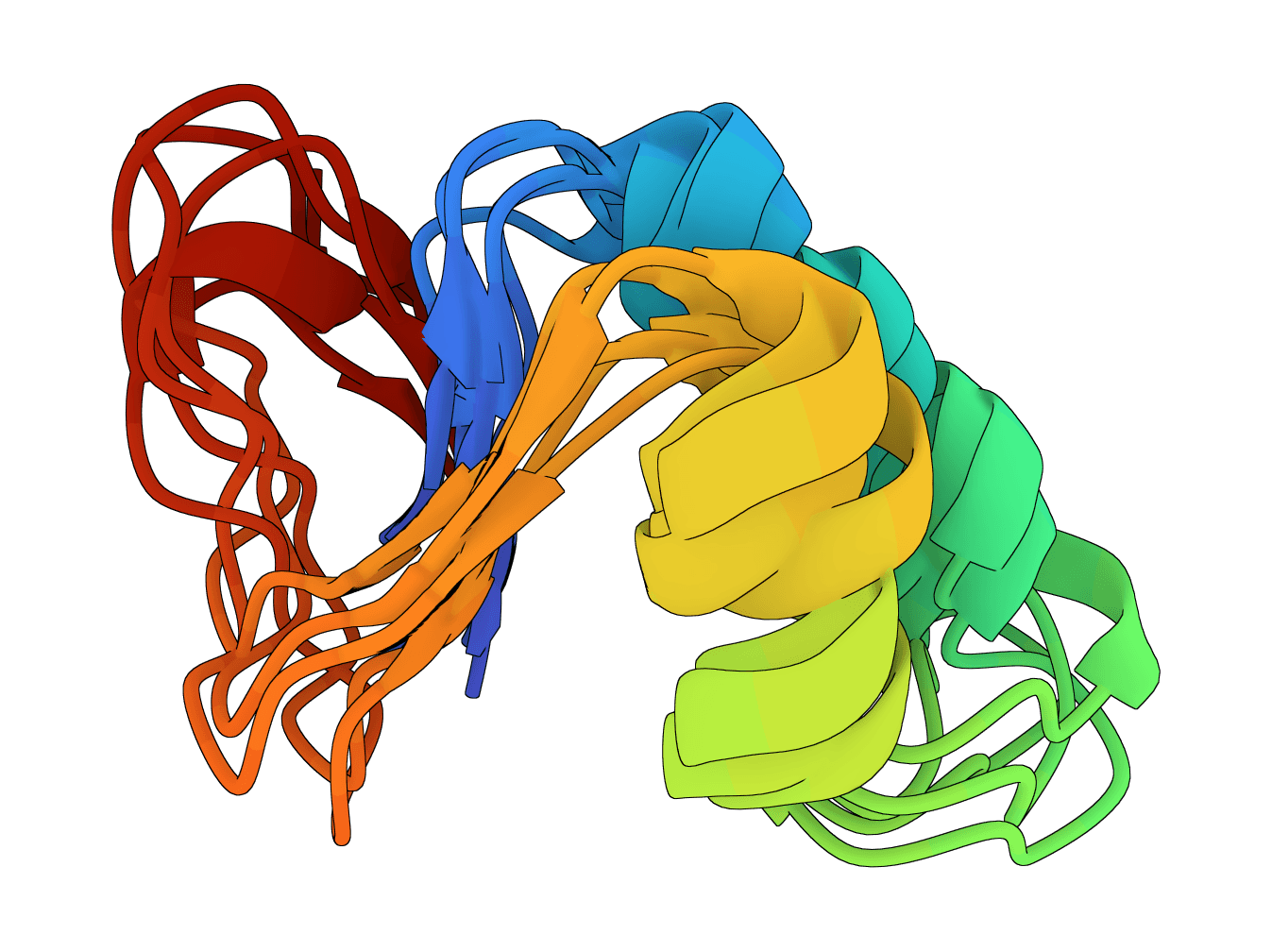
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
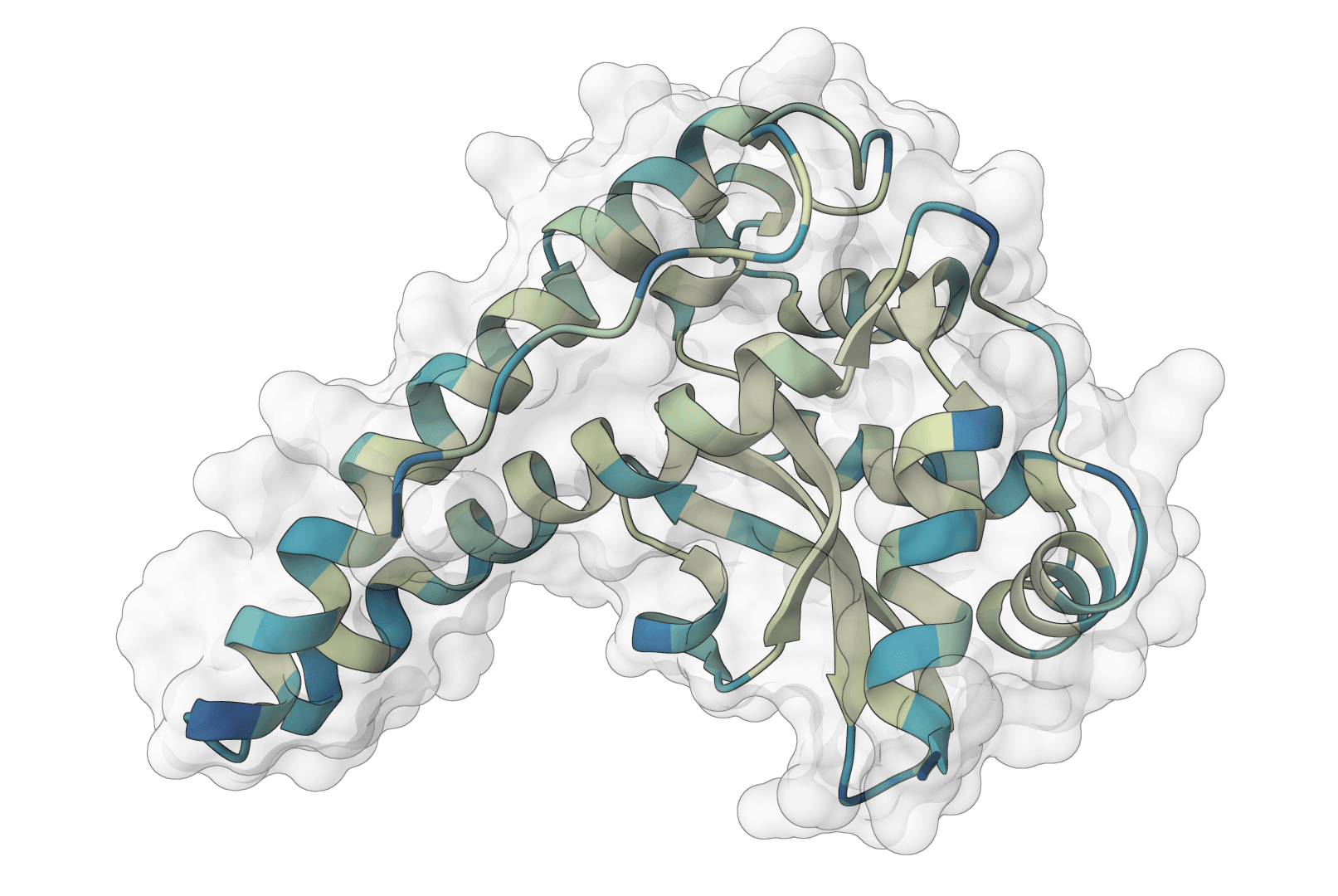
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
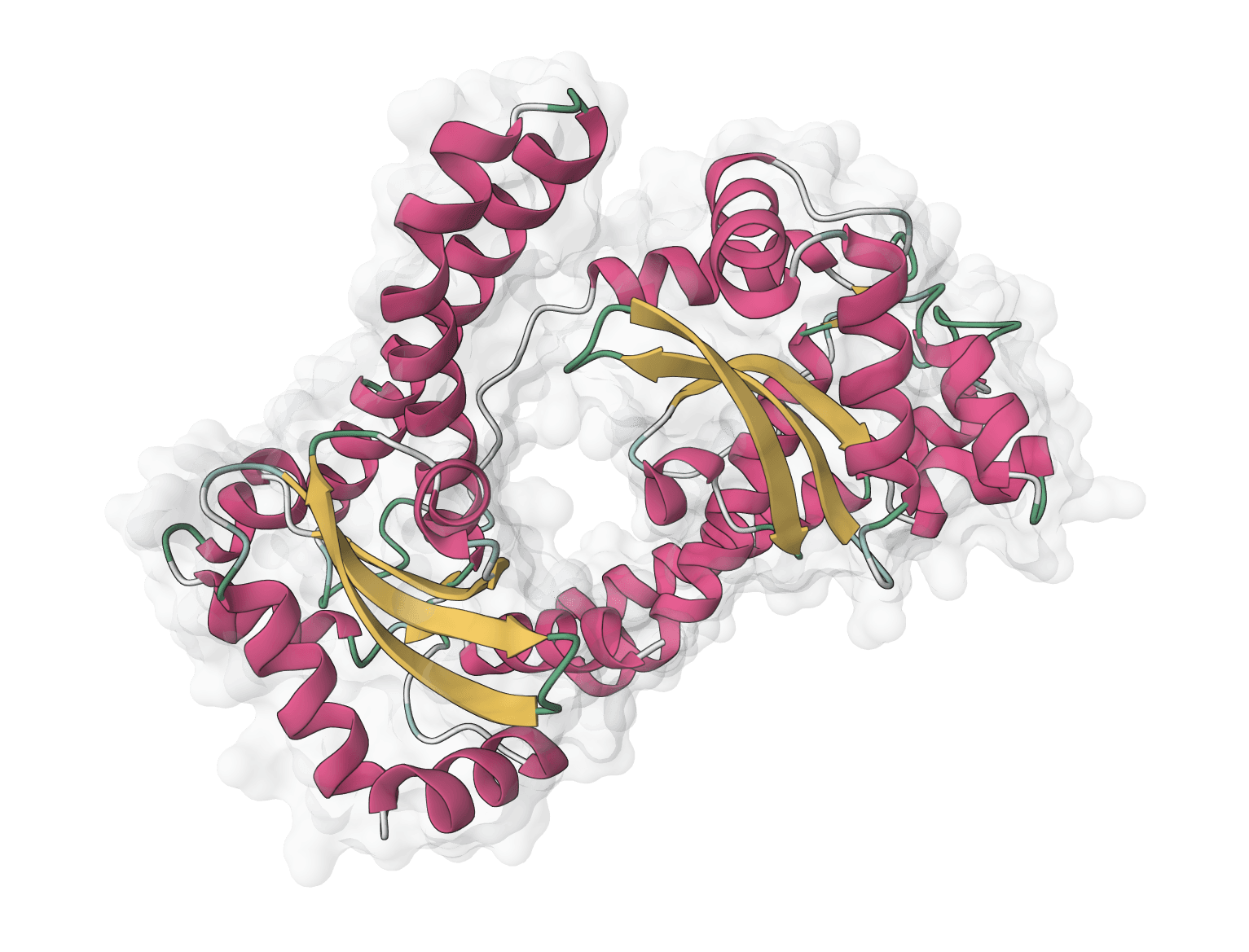
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
IntelliFold 2 is an all-atom biomolecular structure prediction model for proteins, ligands, DNA, RNA, and mixed complexes. It extends the earlier IntFold foundation model with an updated architecture and a faster v2-Flash inference path, while retaining the project's focus on controllable prediction for practical design and screening workflows.
The IntFold technical report describes the platform as a controllable foundation model that reaches AlphaFold 3 level performance on broad biomolecular benchmarks while adding mechanisms for structural constraints, allosteric-state guidance, and downstream ranking. The IntelliFold 2 release positions the newer model family as an architectural refinement intended to improve accuracy on recent FoldBench evaluations.
ProteinIQ provides browser-based access to IntelliFold 2, so the model can be run without local GPU setup, Python installation, or manual environment configuration. Jobs are submitted from a single form that combines molecular inputs, MSA options, and inference settings.
At least one foldable biopolymer is required in practice: a protein, DNA, or RNA chain. The ProteinIQ deployment accepts up to 10 entries for each molecule type, with a combined maximum of 3,000 residues across all protein, DNA, and RNA chains.
| Input | Description |
|---|---|
Protein | FASTA text, .fasta/.fa upload, or an RCSB sequence fetch. Up to 10 chains. |
Precomputed MSA | Optional .a3m or paired .csv alignment files. Up to 10 files. |
Ligand (SMILES) | SMILES entered directly or fetched from PubChem. Up to 10 ligands. |
Ligand (CCD) | PDB Chemical Component Dictionary codes such as ATP, NAD, or SAH. Up to 10 ligands. |
DNA | FASTA text or .fasta/.fa upload. Up to 10 chains. |
RNA | FASTA text or .fasta/.fa upload. Up to 10 chains. |
Job name | Optional label used to identify the run in job history. |
| Setting | Description |
|---|---|
Model | Chooses v2-Flash (default), v2, or v1. v2-Flash is the recommended speed and quality trade-off. |
Diffusion samples | Structures generated per seed (1-10, default 5). Higher values broaden conformational sampling but increase runtime. |
Recycling iterations | Iterative refinement cycles during inference (1-12, default 10). More recycling can improve difficult predictions. |
Sampling steps | Diffusion denoising steps (50-300, default 200). Higher values may improve structure quality at the cost of latency. |
Seeds | One seed or a comma-separated list such as 42,43,44. Multiple seeds produce larger ranked ensembles. |
Output format | mmCIF (default) or PDB. mmCIF is generally preferable for modern structure workflows. |
| Setting | Description |
|---|---|
Generate MSA with server | Uses the MMseqs2 server to build protein MSAs automatically. Disable for single-sequence runs or when uploading precomputed alignments. |
MSA pairing strategy | Pairing mode for multimer alignments when server-side MSA generation is enabled: Greedy (default) or Complete. |
Disable pairing | Turns off paired MSA generation for multimer jobs when only unpaired alignments are desired. |
ProteinIQ returns an interactive structure viewer, a downloadable results table, and the full file bundle for each run.
| Output | Description |
|---|---|
Viewer | Interactive 3D visualization of predicted structures. |
Data table | Ranked prediction summary derived from the primary output files. |
Structure files | Predicted models in mmCIF or PDB, depending on the selected output format. |
Download bundle | All result files produced by the run, including ranked predictions and associated metadata. |
The IntFold project is built as an all-atom foundation model for biomolecular structure prediction. In the authors' description, its distinguishing feature is controllability: the same core system is designed to support general structure prediction as well as guided tasks such as constrained inference, alternative-state modeling, and binding-focused applications.
The IntFold technical report describes specialized adapters that can bias the model toward user-defined structural constraints, allosteric states, and affinity-related downstream tasks. That makes IntelliFold different from sequence-to-structure systems that only return an unconstrained best guess.
ProteinIQ supports the model's diffusion and recycling controls directly. Diffusion sampling starts from noisy coordinates and refines them over many denoising steps, while recycling passes intermediate predictions back through the network for further correction. Increasing either parameter usually improves search depth, but the trade-off is longer runtime.
IntelliFold 2 can use server-generated MSAs through MMseqs2 or run from sequence input with MSA generation disabled. In protein systems with clear evolutionary homologs, MSA depth usually improves structural confidence; disabling MSA is more appropriate for speed-sensitive runs or synthetic targets with weak sequence family coverage.
The IntFold paper also describes a training-free, similarity-based ranking strategy for selecting better predictions from an ensemble. On ProteinIQ, that ensemble is controlled indirectly through Diffusion samples and Seeds, which determine how many candidate structures are generated before ranking.
IntelliFold 2 predictions are typically examined as a ranked set rather than a single definitive structure. Confidence metrics included in the IntelliFold 2 release materials and ProteinIQ configuration are the most useful first-pass filters.
| Metric | Interpretation |
|---|---|
pLDDT | Per-residue confidence on a 0-100 scale. Higher values indicate more reliable local geometry. |
pTM | Predicted template modeling score for overall fold quality. Higher values suggest a more reliable global structure. |
ipTM | Interface confidence for multichain complexes. Most useful when judging protein-protein or protein-nucleic acid assemblies. |
For exploratory jobs, multiple seeds and diffusion samples are often more informative than a single large run with fixed randomness. If top-ranked structures differ substantially from one another, the target may be flexible, weakly constrained by sequence evidence, or genuinely multimodal.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
OpenFold-3 is an open-source AI model for biomolecular structure prediction, aiming to reproduce AlphaFold3. Predicts 3D structures for proteins, RNA, DNA, and small molecule ligands with high accuracy.
Enhanced Protenix v2 biomolecular structure prediction by ByteDance. Predicts 3D structures for proteins, RNA, DNA, and small molecule ligands with high accuracy.
Open-source structure prediction neural network for proteins, nucleic acids, and small molecules. State-of-the-art accuracy with multi-chain support.
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
ABodyBuilder3 predicts antibody variable-domain structures from paired heavy and light chain sequences. It returns a PDB structure and, for the pLDDT checkpoint, per-residue confidence values.
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.