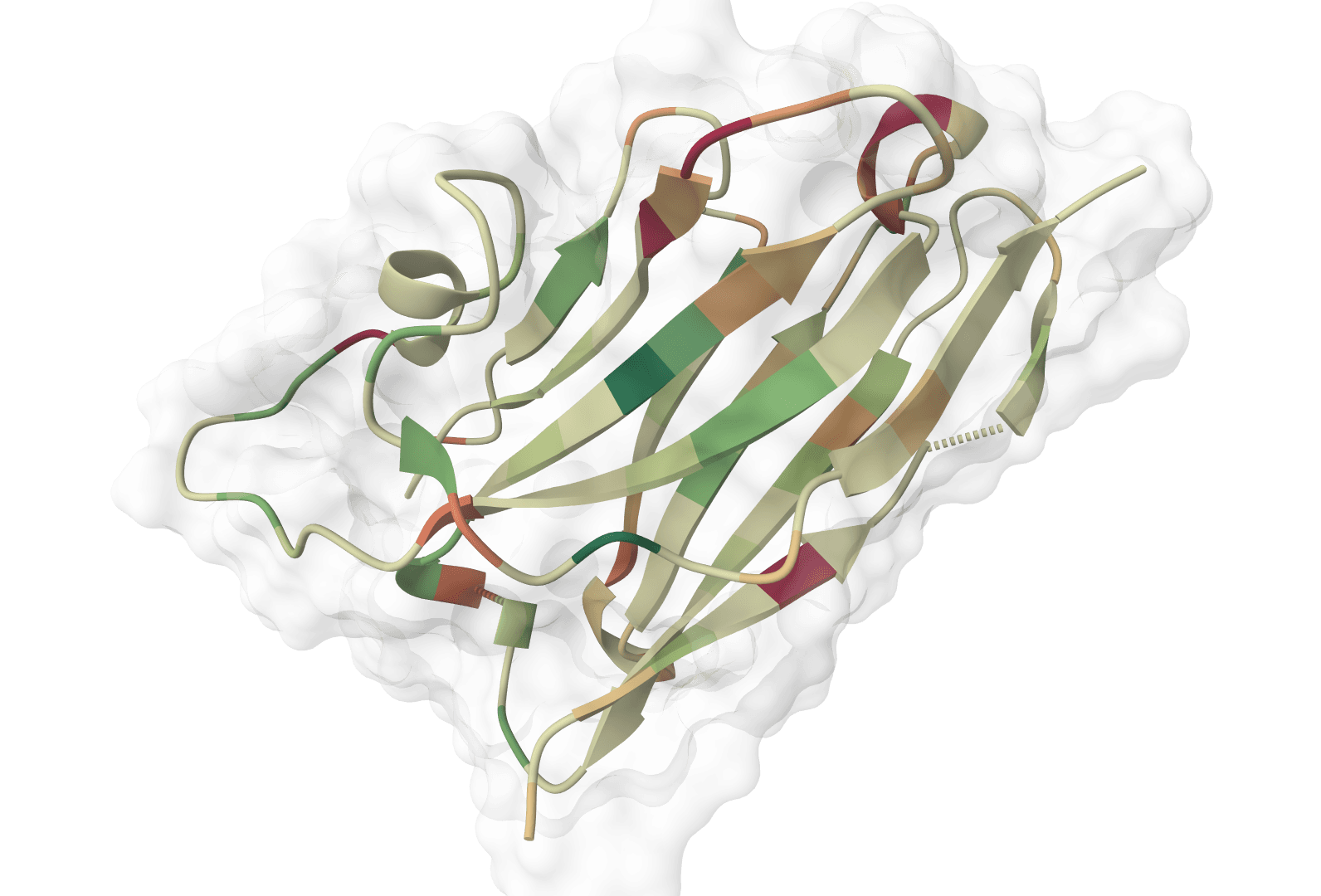
ABodyBuilder3 predicts the 3D structure of an antibody variable domain (Fv) from its paired heavy and light chain sequences. It is the third generation of the ABodyBuilder line from the Oxford Protein Informatics Group, built on the ImmuneBuilder framework, and it specializes in the part of antibody modeling that general structure predictors handle worst: the hypervariable CDR loops, especially CDR-H3.
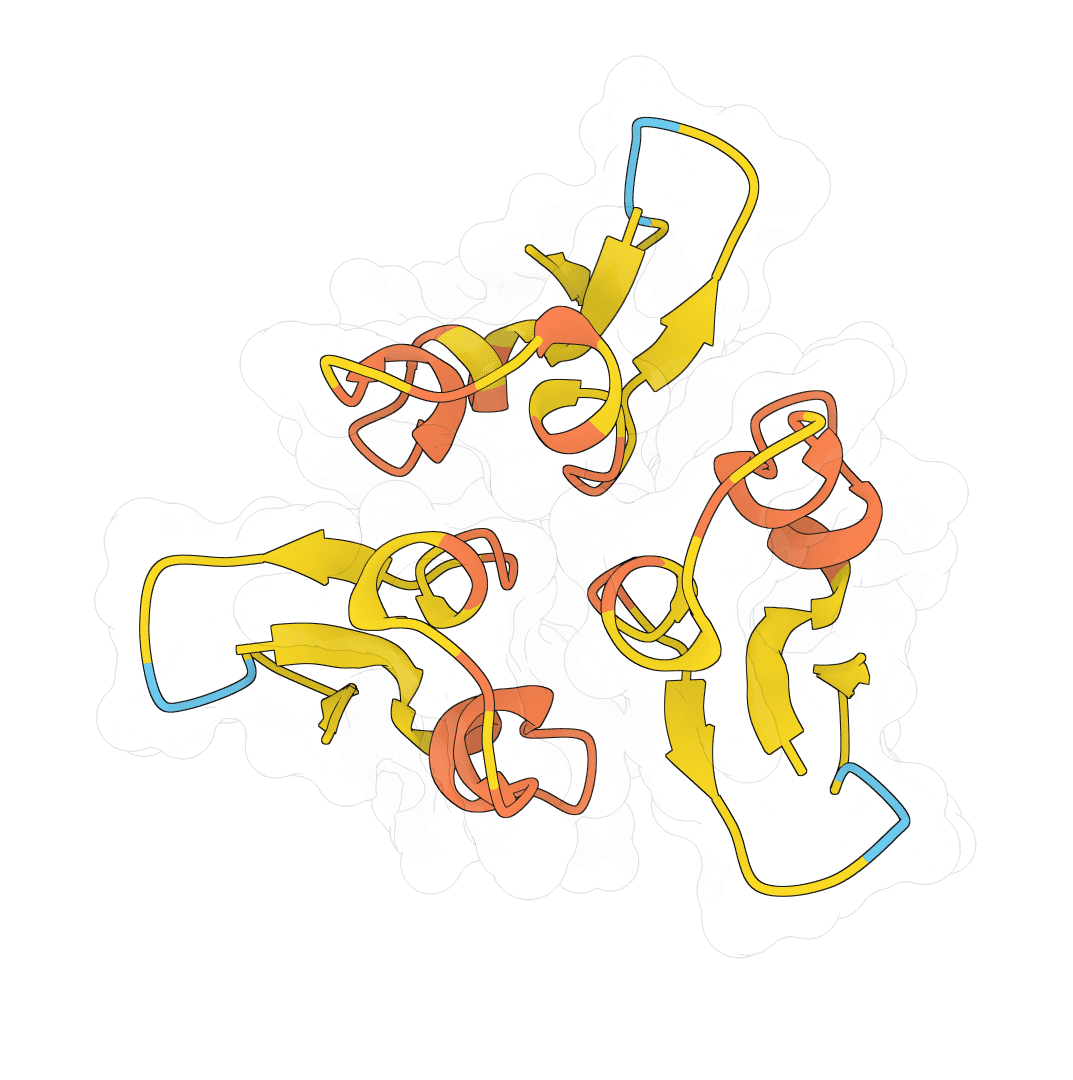
Most of an antibody Fv is conformationally conserved, so the framework regions are easy to model. The binding loops are the hard part. CDR-H3 has no canonical templates, varies in length and sequence across antibodies, and largely determines where and how the antibody binds. ABodyBuilder3 focuses its accuracy there.
Paste an antibody heavy chain (VH) and light chain (VL) sequence into ProteinIQ, pick a model checkpoint, and run the prediction on cloud GPUs. The result is a single Fv structure in PDB format with heavy and light written as separate chains, plus a summary of chain lengths and, on the pLDDT checkpoint, per-residue confidence. No installation, no antibody numbering setup, and no MSA step.
| Input | Format | Notes |
|---|---|---|
Heavy chain (VH) | Amino-acid sequence or FASTA | Standard 20-letter alphabet; X is accepted for an unknown residue. Provide the variable domain only, not the full heavy chain. |
Light chain (VL) | Amino-acid sequence or FASTA | Required. ABodyBuilder3 models paired Fv, so both chains are needed. |
Both inputs expect the variable domain sequence. Trimming constant-region residues before submitting avoids modeling regions the tool is not trained on. To assign and trim variable domains by antibody numbering, use ANARCI.
| Setting | Default | Description |
|---|---|---|
Model checkpoint | ABodyBuilder3-LM loss checkpoint | Selects which trained model runs. The LM checkpoint uses protein language model embeddings for residue representation. The pLDDT checkpoint additionally predicts per-residue confidence. |
Two checkpoints are available because the paper trains the model two ways:
ABodyBuilder3-LM loss checkpoint: the default. Selected on validation loss, it gives the most accurate backbone for routine structure prediction.ABodyBuilder3 pLDDT checkpoint: trained with a confidence head that emits per-residue pLDDT. Choose this when the confidence values matter, for example to flag an unreliable CDR-H3 loop before downstream docking.| Output | Format | Notes |
|---|---|---|
| Predicted Fv structure | PDB | Heavy and light domains as separate chains. |
| pLDDT data | JSON | Returned only on the pLDDT checkpoint. |
| Summary table | Spreadsheet | Heavy and light chain lengths, total residues, model checkpoint, and mean pLDDT when available. |
ABodyBuilder3 inherits the ImmuneBuilder architecture: a graph-based network predicts inter-residue geometry, which is folded into atomic coordinates and refined. The two changes that define this version are the sequence representation and the confidence estimate.
ABodyBuilder2 encoded each residue as a one-hot vector. ABodyBuilder3 replaces that with embeddings from the ProtT5 protein language model, which has seen billions of natural protein sequences and carries evolutionary and structural context that a one-hot vector cannot. This richer representation stabilizes training and improves modeling of the variable loops, with the largest effect on CDR-H3 and CDR-L3. The gain on CDR-H3 backbone RMSD is real but modest; the more reliable benefit is more robust training and a model that no longer needs an ensemble to estimate confidence.
Earlier ImmuneBuilder models estimated per-residue uncertainty by training an ensemble of networks and measuring their disagreement, which multiplies both training and inference cost. ABodyBuilder3 adds a pLDDT head that predicts local accuracy directly, in the same 0 to 100 scale used by AlphaFold. A single model with this head matches the ensemble's uncertainty estimate at a fraction of the compute.
| pLDDT | Meaning |
|---|---|
| > 90 | High confidence, backbone very likely correct. |
| 70 to 90 | Reliable, framework and well-defined loops. |
| 50 to 70 | Treat with caution, common in long or unusual CDR-H3 loops. |
| < 50 | Low confidence, the local geometry may be wrong. |
Confidence tends to be high across the framework and lower through the CDRs. A low score concentrated in CDR-H3 is expected for long or atypical loops and is the signal to validate that region before relying on it.
ABodyBuilder3 is the right choice when the target is a paired antibody Fv and the goal is a fast, accurate model of the binding loops. It is purpose-built for paired VH/VL variable domains, runs in seconds, and needs no MSA. For a single-chain nanobody, use ImmuneBuilder with NanoBodyBuilder2.
For a full antigen-antibody complex rather than the unbound Fv, a co-folding model such as Chai-1 or Protenix predicts antibody and antigen together. For general (non-antibody) single-chain prediction, use ESMFold or AlphaFold 2. To redesign CDR sequences or graft new loops onto a predicted scaffold, pair the structure with AntiFold, Proteo-R1, or the antibody-specialized RFantibody and DiffAb.
A typical workflow numbers and trims the chains with ANARCI, predicts the Fv here, then docks against an antigen or feeds the structure into a design step. To compare a prediction against a crystal structure or another model, measure backbone agreement with the RMSD calculator.
ImmuneBuilder predicts 3D structures of immune receptor proteins including antibodies, nanobodies, and T-cell receptors. It uses ABodyBuilder2, NanoBodyBuilder2, and TCRBuilder2/TCRBuilder2+ to generate structures with per-residue error estimates and optional ensemble artifacts.
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
ESMFold2 predicts protein structures and multi-chain protein complexes from amino acid sequences using Biohub protein language models. The first ProteinIQ release focuses on sequence-based protein folding with confidence metrics, native mmCIF structures, and optional PAE and pair-chain iPTM outputs.
Controllable biomolecular structure prediction model for proteins, ligands, DNA, RNA, and multi-component complexes. IntelliFold 2 supports fast v2-Flash inference, optional MSA generation, and ranked confidence outputs.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
MiniFold is a fast single-sequence protein structure predictor that is 10-20x faster than ESMFold. It predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it ideal for rapid structure prediction.
ABodyBuilder3 predicts the 3D structure of an antibody variable domain (Fv) from its paired heavy and light chain sequences. It is the third generation of the ABodyBuilder line from the Oxford Protein Informatics Group, built on the ImmuneBuilder framework, and it specializes in the part of antibody modeling that general structure predictors handle worst: the hypervariable CDR loops, especially CDR-H3.
Most of an antibody Fv is conformationally conserved, so the framework regions are easy to model. The binding loops are the hard part. CDR-H3 has no canonical templates, varies in length and sequence across antibodies, and largely determines where and how the antibody binds. ABodyBuilder3 focuses its accuracy there.
Paste an antibody heavy chain (VH) and light chain (VL) sequence into ProteinIQ, pick a model checkpoint, and run the prediction on cloud GPUs. The result is a single Fv structure in PDB format with heavy and light written as separate chains, plus a summary of chain lengths and, on the pLDDT checkpoint, per-residue confidence. No installation, no antibody numbering setup, and no MSA step.
| Input | Format | Notes |
|---|---|---|
Heavy chain (VH) | Amino-acid sequence or FASTA | Standard 20-letter alphabet; X is accepted for an unknown residue. Provide the variable domain only, not the full heavy chain. |
Light chain (VL) | Amino-acid sequence or FASTA | Required. ABodyBuilder3 models paired Fv, so both chains are needed. |
Both inputs expect the variable domain sequence. Trimming constant-region residues before submitting avoids modeling regions the tool is not trained on. To assign and trim variable domains by antibody numbering, use ANARCI.
| Setting | Default | Description |
|---|---|---|
Model checkpoint | ABodyBuilder3-LM loss checkpoint | Selects which trained model runs. The LM checkpoint uses protein language model embeddings for residue representation. The pLDDT checkpoint additionally predicts per-residue confidence. |
Two checkpoints are available because the paper trains the model two ways:
ABodyBuilder3-LM loss checkpoint: the default. Selected on validation loss, it gives the most accurate backbone for routine structure prediction.ABodyBuilder3 pLDDT checkpoint: trained with a confidence head that emits per-residue pLDDT. Choose this when the confidence values matter, for example to flag an unreliable CDR-H3 loop before downstream docking.| Output | Format | Notes |
|---|---|---|
| Predicted Fv structure | PDB | Heavy and light domains as separate chains. |
| pLDDT data | JSON | Returned only on the pLDDT checkpoint. |
| Summary table | Spreadsheet | Heavy and light chain lengths, total residues, model checkpoint, and mean pLDDT when available. |
ABodyBuilder3 inherits the ImmuneBuilder architecture: a graph-based network predicts inter-residue geometry, which is folded into atomic coordinates and refined. The two changes that define this version are the sequence representation and the confidence estimate.
ABodyBuilder2 encoded each residue as a one-hot vector. ABodyBuilder3 replaces that with embeddings from the ProtT5 protein language model, which has seen billions of natural protein sequences and carries evolutionary and structural context that a one-hot vector cannot. This richer representation stabilizes training and improves modeling of the variable loops, with the largest effect on CDR-H3 and CDR-L3. The gain on CDR-H3 backbone RMSD is real but modest; the more reliable benefit is more robust training and a model that no longer needs an ensemble to estimate confidence.
Earlier ImmuneBuilder models estimated per-residue uncertainty by training an ensemble of networks and measuring their disagreement, which multiplies both training and inference cost. ABodyBuilder3 adds a pLDDT head that predicts local accuracy directly, in the same 0 to 100 scale used by AlphaFold. A single model with this head matches the ensemble's uncertainty estimate at a fraction of the compute.
| pLDDT | Meaning |
|---|---|
| > 90 | High confidence, backbone very likely correct. |
| 70 to 90 | Reliable, framework and well-defined loops. |
| 50 to 70 | Treat with caution, common in long or unusual CDR-H3 loops. |
| < 50 | Low confidence, the local geometry may be wrong. |
Confidence tends to be high across the framework and lower through the CDRs. A low score concentrated in CDR-H3 is expected for long or atypical loops and is the signal to validate that region before relying on it.
ABodyBuilder3 is the right choice when the target is a paired antibody Fv and the goal is a fast, accurate model of the binding loops. It is purpose-built for paired VH/VL variable domains, runs in seconds, and needs no MSA. For a single-chain nanobody, use ImmuneBuilder with NanoBodyBuilder2.
For a full antigen-antibody complex rather than the unbound Fv, a co-folding model such as Chai-1 or Protenix predicts antibody and antigen together. For general (non-antibody) single-chain prediction, use ESMFold or AlphaFold 2. To redesign CDR sequences or graft new loops onto a predicted scaffold, pair the structure with AntiFold, Proteo-R1, or the antibody-specialized RFantibody and DiffAb.
A typical workflow numbers and trims the chains with ANARCI, predicts the Fv here, then docks against an antigen or feeds the structure into a design step. To compare a prediction against a crystal structure or another model, measure backbone agreement with the RMSD calculator.
ImmuneBuilder predicts 3D structures of immune receptor proteins including antibodies, nanobodies, and T-cell receptors. It uses ABodyBuilder2, NanoBodyBuilder2, and TCRBuilder2/TCRBuilder2+ to generate structures with per-residue error estimates and optional ensemble artifacts.
Generate protein conformational ensembles with ESMFlow, the single-sequence AlphaFlow model family. Produces multiple diverse structures showing protein flexibility and dynamics.
AlphaFold2 via ColabFold for protein structure prediction. Free runs use single-sequence mode; paid plans add MMseqs2 MSA generation. Supports monomer and multimer prediction.
Boltz-2 is a biomolecular foundation model for structure and binding affinity prediction. Supports proteins, ligands, DNA, and RNA in multi-component complexes. Automatically scales GPU resources for large complexes. Predicts binding affinity with near-FEP accuracy at 1000x faster speed.
Chai-1 is a multi-modal foundation model for molecular structure prediction. Predicts 3D structures for proteins, ligands, DNA, RNA, and multi-component complexes with high accuracy.
ESMfold is a fast, single-sequence protein structure predictor from Meta AI. Predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it significantly faster than AlphaFold while automatically scaling GPU resources for larger proteins.
ESMFold2 predicts protein structures and multi-chain protein complexes from amino acid sequences using Biohub protein language models. The first ProteinIQ release focuses on sequence-based protein folding with confidence metrics, native mmCIF structures, and optional PAE and pair-chain iPTM outputs.
Controllable biomolecular structure prediction model for proteins, ligands, DNA, RNA, and multi-component complexes. IntelliFold 2 supports fast v2-Flash inference, optional MSA generation, and ranked confidence outputs.
LMI4Boltz is a low-memory fork of Boltz for biomolecular structure and binding affinity prediction. It preserves Boltz inference behavior while reducing VRAM use with in-place pair updates, CPU offload, reduced precision pair representation, and aggressive chunking.
MiniFold is a fast single-sequence protein structure predictor that is 10-20x faster than ESMFold. It predicts 3D protein structures directly from amino acid sequences without requiring multiple sequence alignments (MSA), making it ideal for rapid structure prediction.
