Introducing DiffDock-L on ProteinIQ

We've integrated DiffDock-L, a state-of-the-art molecular docking tool that predicts protein-ligand binding poses using diffusion models—the same AI architecture that powers modern image generation.
Molecular docking is a foundational step in drug discovery. Understanding how small molecules bind to protein targets guides lead optimization, virtual screening campaigns, and structure-based design. But running these tools has always required technical expertise, including managing dependencies, configuring GPU clusters, and troubleshooting installation failures.
ProteinIQ makes DiffDock-L easily accessible—you open your browser, upload a protein structure and ligand, and get docking predictions in roughly a minute.
Performance and infrastructure
Our implementation runs on NVIDIA A100 GPUs—professional-grade compute infrastructure that delivers results in roughly a minute. The model generates multiple binding poses with confidence scores, typically completing analysis while you're still reviewing your input files.
Many platforms offering molecular docking require 5-10 minutes of compute time. Our A100 infrastructure handles the same workload ten times faster without compromising accuracy.
Why DiffDock-L
We chose to integrate DiffDock-L rather than the original DiffDock because it represents a meaningful step forward in accuracy. On standard benchmarks, DiffDock-L achieves a 43% success rate for poses within 2Å RMSD, compared to 38% for the original DiffDock. This improvement is particularly notable for flexible ligands and challenging binding sites.
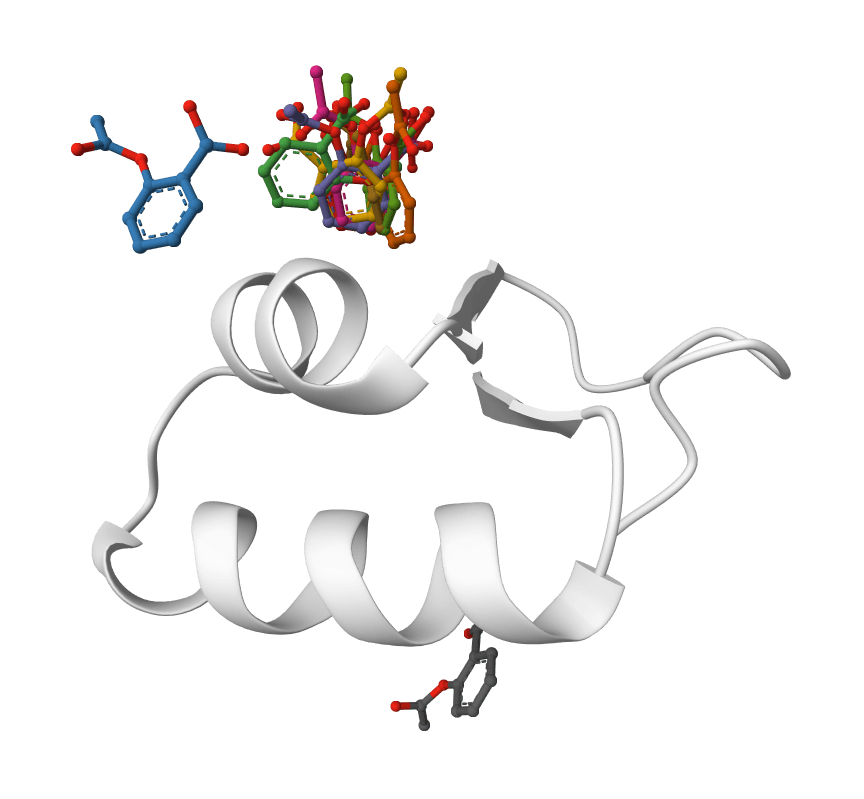
The diffusion model approach also outperforms traditional force-field-based docking methods, especially for complex binding modes that require exploring diverse conformational space.
The tool accepts:
-
Protein structures as PDB files or RCSB PDB IDs
-
Ligands as SMILES strings or SDF files
-
Multiple poses per complex (configurable 1-40)
-
Customizable inference parameters for speed/accuracy tradeoffs Each prediction includes:
-
Ranked binding poses with confidence scores
-
3D structure visualization in the browser
-
Downloadable coordinate files for further analysis
-
Full parameter logging for reproducibility
Getting started
DiffDock-L is available now on ProteinIQ. If you're using the platform, navigate to Prediction → DiffDock-L to start docking. Free tier users can run up to 20 jobs to explore the tool's capabilities.
For typical usage:
- Upload or fetch your protein structure (PDB format)
- Provide your ligand (SMILES or SDF)
- Configure poses and inference steps (defaults work well)
- Submit and review results in ~1 minute The model was trained on diverse protein-ligand complexes and generalizes well to unseen targets. For optimal results, ensure your protein structure is prepared (missing residues fixed, appropriate protonation states) and your ligand is a reasonable drug-like molecule.
What this enables
Fast, accurate docking means:
- Rapid iteration: Test binding hypotheses quickly during lead optimization
- Accessible screening: Run small virtual screens without cluster access
- Educational use: Students learn docking without installation barriers
- Collaborative research: Share docking setups and results with team members seamlessly
Further information
DiffDock-L was developed by the Coley Lab at MIT and is based on the research published in "DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking". The model architecture uses SE(3) equivariant diffusion to predict ligand poses while respecting the 3D symmetries inherent to molecular systems. We've integrated this model into our platform with optimized infrastructure for fast, accessible execution.
For technical details, see the DiffDock-L tool page. For questions about infrastructure, pricing, or custom deployments, contact our team.
Every researcher deserves access to the tools that accelerate discovery. We're here to make that happen.