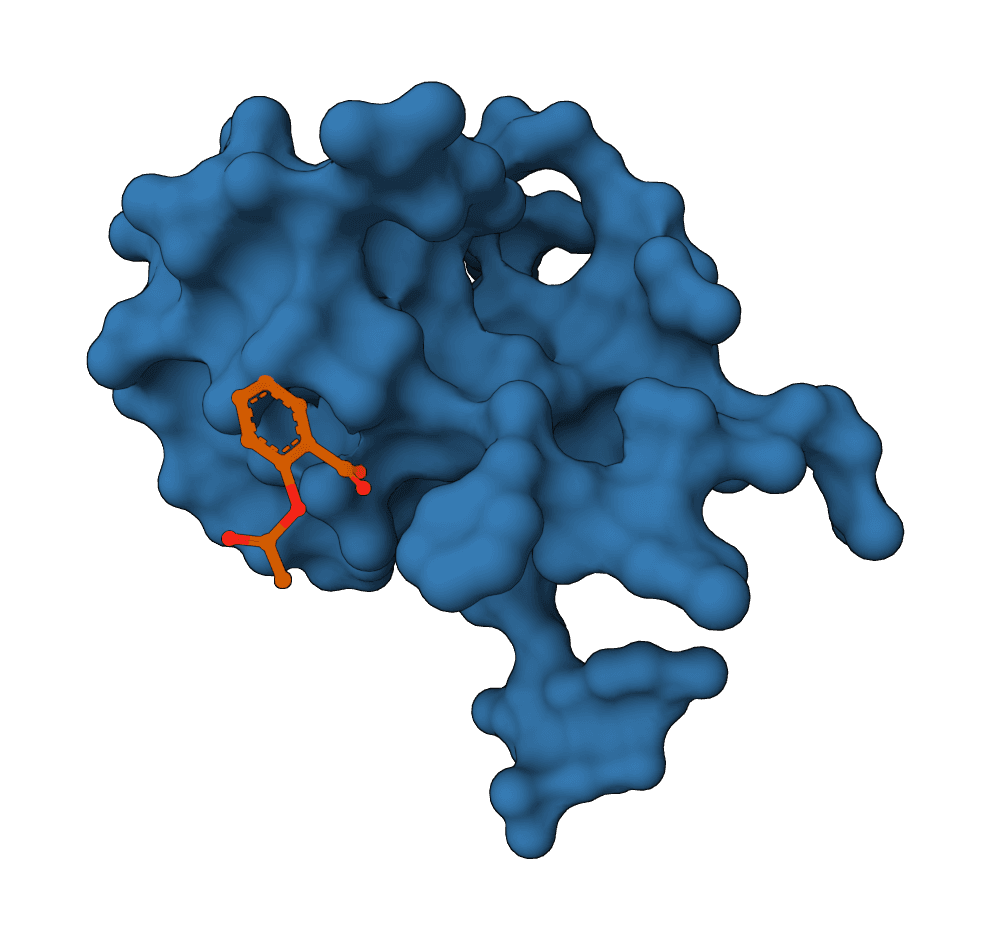
Geometric flow matching for generative protein-ligand docking and affinity prediction. Learn more
Run
75 credits
Output
Configure inputs to begin
Set options on the left, then click “Submit job”.
Geometric flow matching for generative protein-ligand docking and affinity prediction. Learn more
Configure inputs to begin
Set options on the left, then click “Submit job”.
Geometric flow matching for generative protein-ligand docking and affinity prediction. Learn more
Configure inputs to begin
Set options on the left, then click “Submit job”.
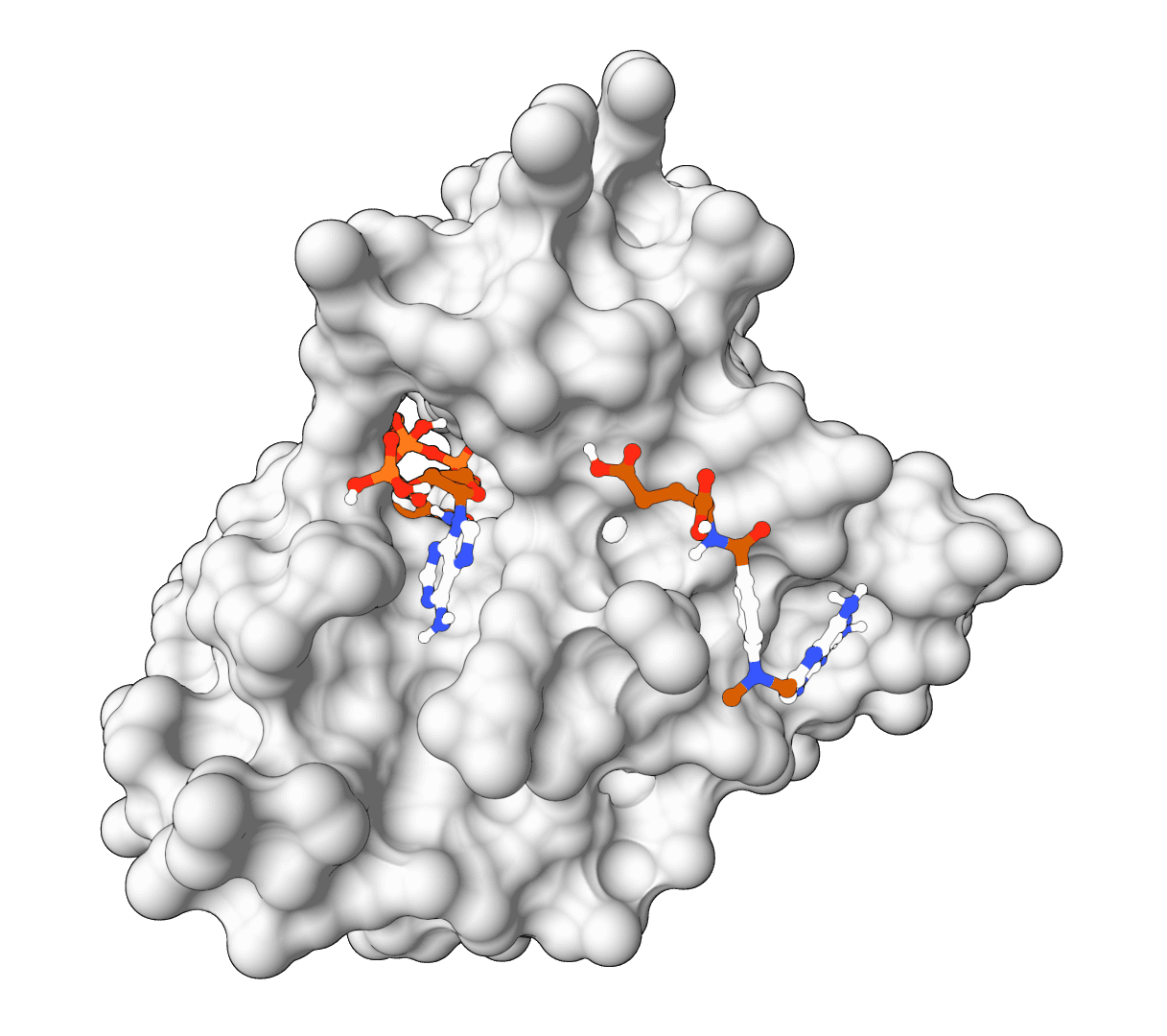
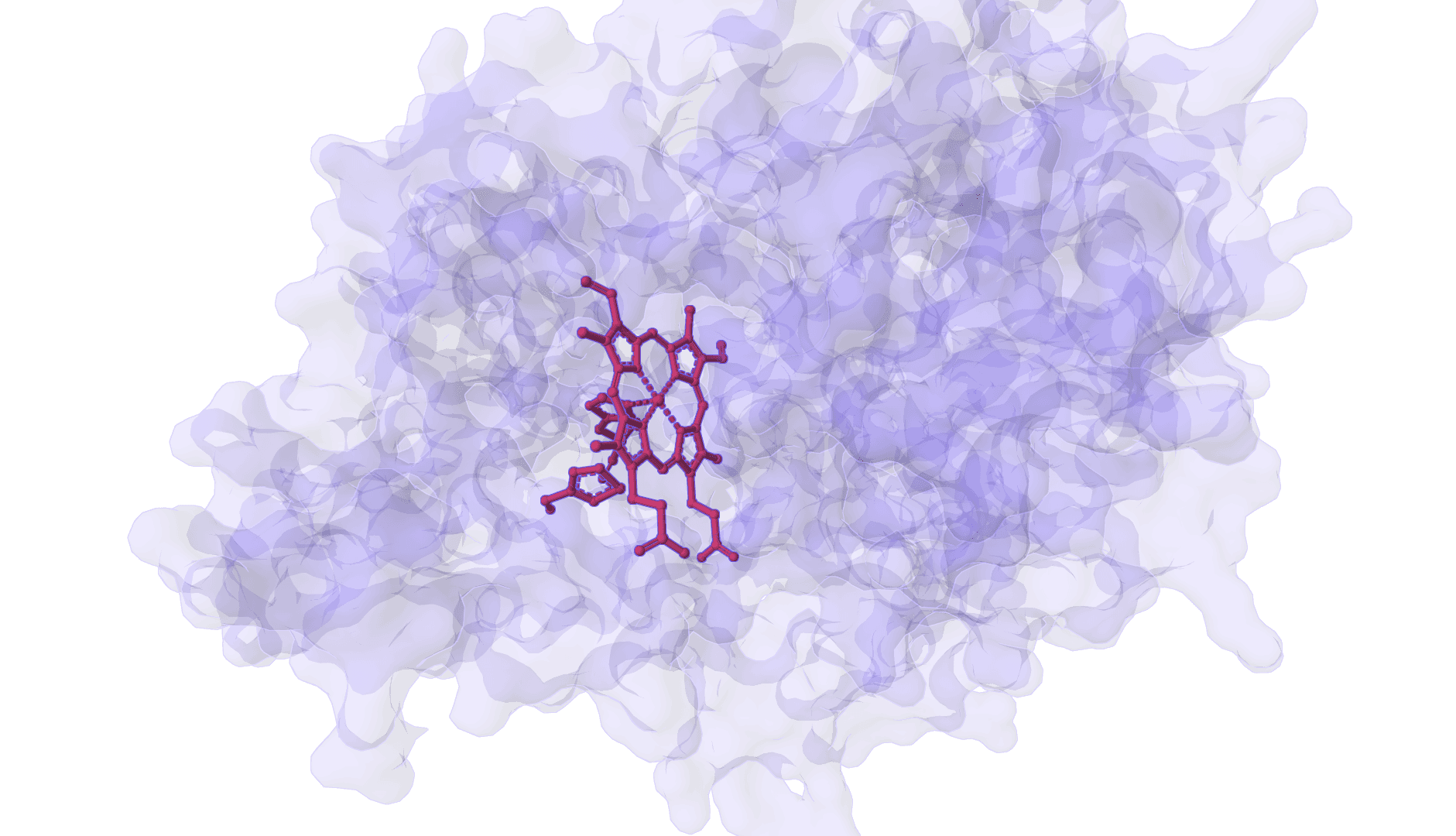
FlowDock predicts the structure of a protein-ligand complex and estimates how tightly the two bind, starting from nothing more than a protein sequence and a ligand SMILES string. It is a generative model: instead of slotting a ligand into a fixed receptor, it builds the bound protein conformation and the docked ligand pose together, then reports a confidence score and an affinity estimate for each sampled complex.
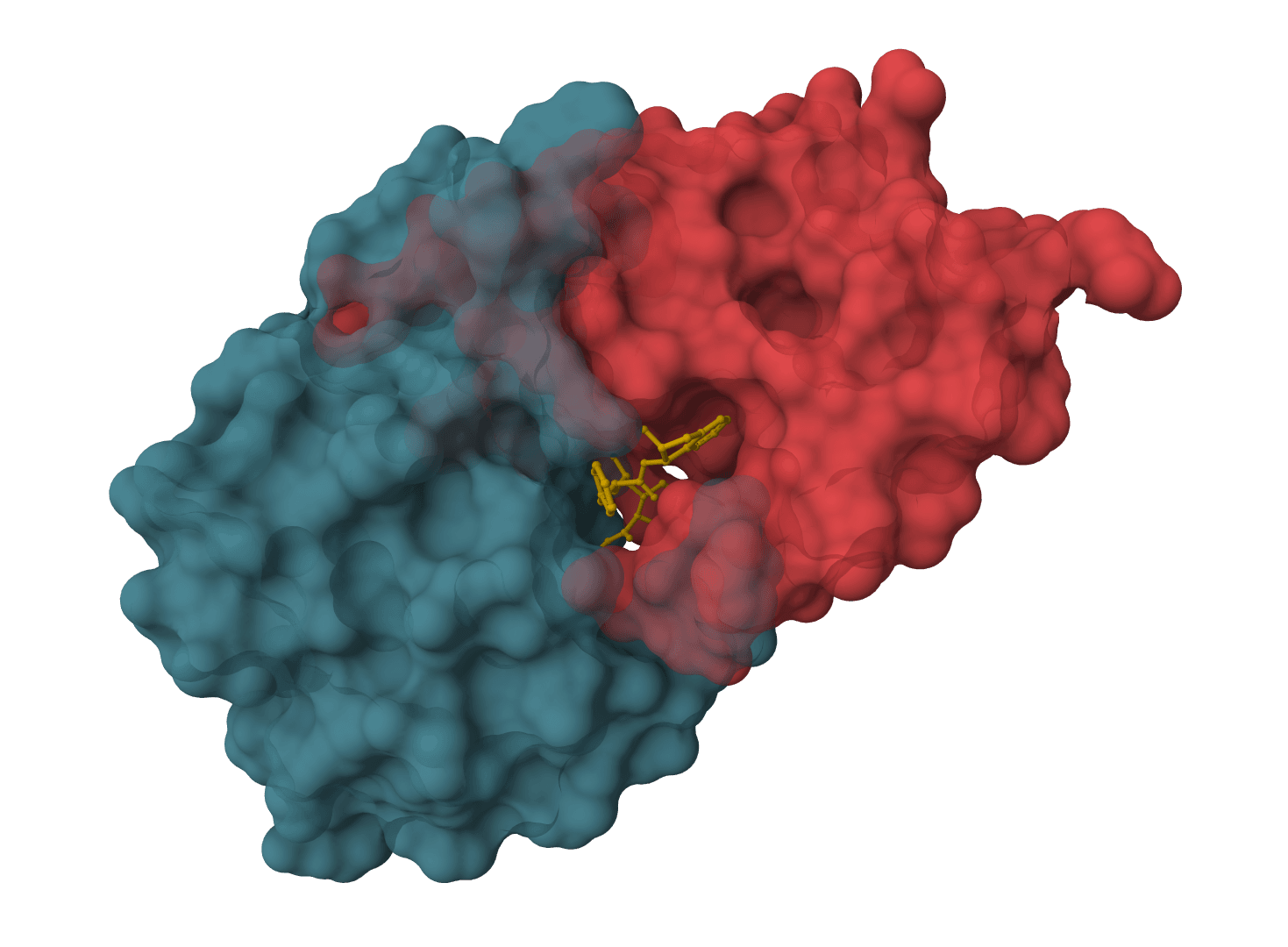
That single trait separates it from most docking tools. Classic docking assumes a known receptor structure and treats the protein as rigid or nearly so. FlowDock generates the holo (bound) protein from an apo prior, so the backbone and side chains can move to accommodate the ligand. The cost of that flexibility is sampling: FlowDock draws several complexes per run and ranks them, rather than returning a single deterministic answer.
The method comes from Alex Morehead and Jianlin Cheng (ISMB 2025). It is most useful when no experimental holo structure exists, when binding may reshape the pocket, or when a fast affinity estimate alongside the pose is more valuable than a single high-resolution docking result.
ProteinIQ runs FlowDock on GPU infrastructure with no setup. Provide a protein as a sequence, FASTA, or receptor PDB, and a ligand as a SMILES string or SDF file. FlowDock samples several protein-ligand complexes and returns ranked PDB structures, ranked SDF ligand poses, per-pose plDDT confidence, and an affinity estimate for each, all downloadable. An optional template PDB can guide the structure prior.
| Input | Supported formats | Notes |
|---|---|---|
Protein | Plain sequence, FASTA, or PDB | Up to 4096 residues. Separate sequence chains with |. A PDB is used directly as the receptor. |
Ligand | SMILES or SDF | Up to 300 heavy atoms. Use | between SMILES strings for multi-ligand inputs. |
Protein template | PDB (optional) | Used by the ESMFold structure prior when it matches the input sequence. |
Two ProteinIQ tools pair naturally with the inputs here. If only a ligand name or PubChem CID is on hand, fetch the SMILES directly in the ligand card. If the protein is known only by sequence and a starting fold helps, predict one first with ESMFold and pass it as the template.
FlowDock's defaults reproduce the sampling configuration from the original sample.py, so the out-of-the-box run matches the published method. The settings below mostly trade speed against the number and diversity of sampled complexes.
| Setting | Description |
|---|---|
Number of samples | Complexes to generate (1-20, default 5). More samples explore more poses but take longer. Keep it divisible by chunk size. |
Chunk size | Samples processed per batch (1-10, default 5). Number of samples should divide evenly by this value. |
Sampling steps | Integration steps along the flow (10-80, default 40). More steps refine geometry at the cost of runtime. |
Sampler | VDODE (default) or ODE. The variance-diminishing ODE sampler is the published default. |
Sampler eta | Noise scale for the sampler (0.5-2.0, default 1.0). Lower values are more deterministic. |
Start time | 1.0 (default) starts from the full prior; auto lets FlowDock pick the start point. |
Use template | Applies the supplied template PDB to the structure prior when the sequence matches (default on). |
Discard SDF coordinates | Ignores input ligand coordinates and samples poses from scratch (default on). |
Rank by confidence | Orders outputs by the confidence head and writes ranked files (default on). |
Ranking score | Which plDDT drives ranking: Ligand (default), Protein, or Protein + ligand. Ligand plDDT is the right choice when pose quality matters most. |
| Output | Description |
|---|---|
prot_all.pdb | Multi-model PDB with every sampled protein structure. |
lig_all.sdf | Multi-conformer SDF with every sampled ligand pose. |
prot_rank*.pdb | Ranked protein structures (when confidence ranking is on). |
lig_rank*.sdf | Ranked ligand poses (when confidence ranking is on). |
lig_ref.sdf | Reference ligand SDF, written when ligand coordinates are available. |
*_auxiliary_estimation.csv | Per-pose rank, plDDT values, and affinity estimates. |
The Data tab summarizes the same results as a sortable table of rank, plDDT, affinity, and source filename, while the Viewer tab loads the sampled complexes for inspection.
FlowDock treats docking as a transport problem. It learns a continuous flow that carries an unbound starting structure toward a bound protein-ligand complex, parameterized by conditional flow matching. At inference it integrates that flow over a fixed number of steps, moving atoms from the prior toward a plausible holo state.
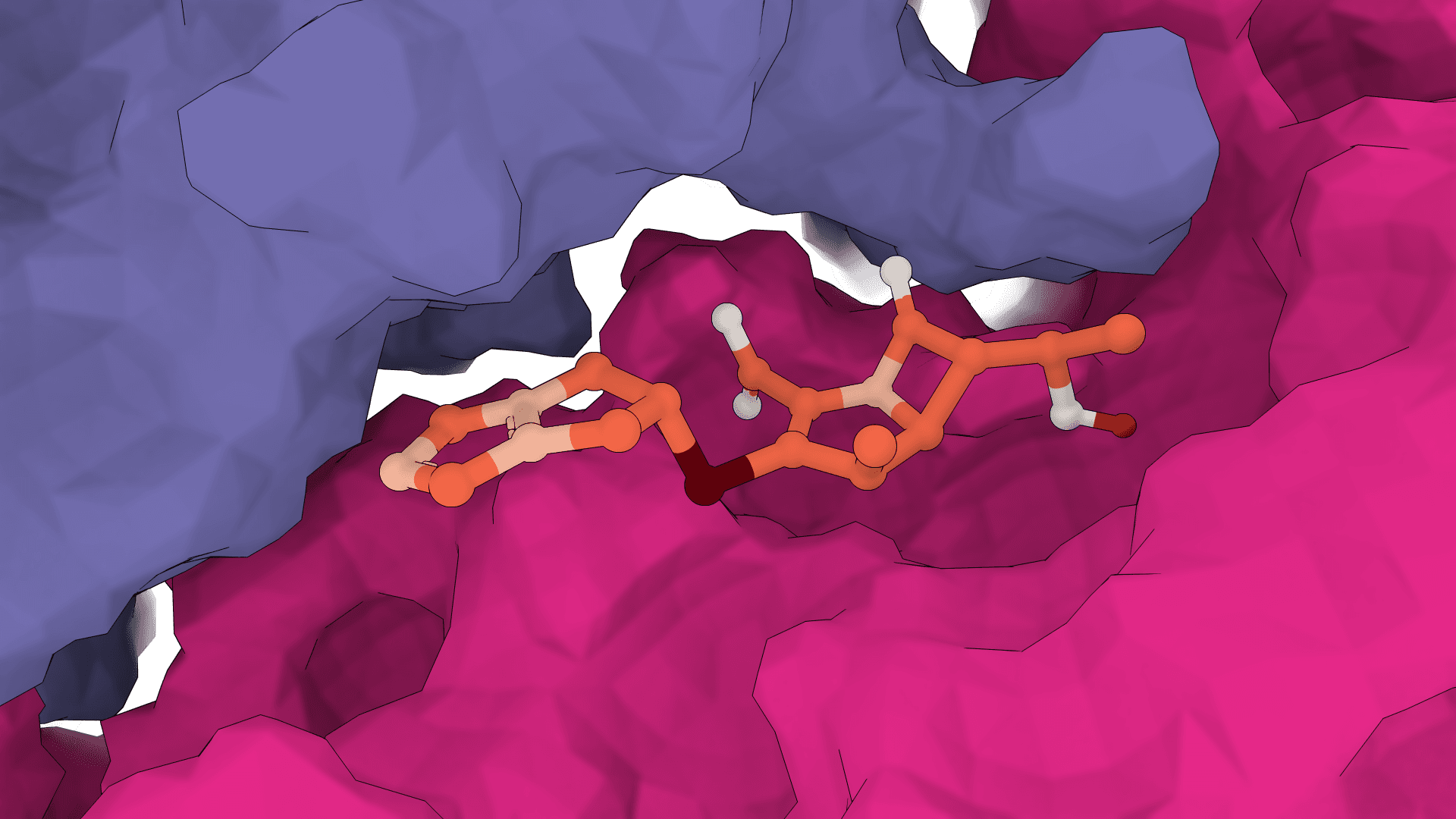
The starting point comes from a structure prior. FlowDock uses an ESMFold-derived apo conformation of the protein, which is why a sequence is enough to start and why a matching template PDB can be supplied to steer the prior. The ligand is initialized from its molecular graph, with input coordinates discarded by default so the pose is sampled rather than copied.
Two auxiliary heads run on top of the generated geometry. A confidence head produces plDDT-style per-residue and per-ligand scores, and an affinity head predicts binding strength. Because generation is stochastic, FlowDock draws several complexes and uses the confidence head to rank them, so the top-ranked file is the model's best guess rather than the only one it considered.
plDDT here mirrors the confidence metric from structure prediction: higher means the model is more certain about the local geometry it produced. Ligand plDDT speaks to pose reliability, protein plDDT to the backbone and pocket. Treat a high-plDDT top pose as a confident prediction and a run where even the best pose scores low as a signal that the pocket, the ligand, or the prior is hard for the model.
The affinity values are model estimates, not measurements. Use them to rank candidates against each other within a campaign, not as a substitute for an assay or a physics-based free energy. When ranking matters, compare relative affinity across ligands docked the same way rather than reading any single number as an absolute Kd.
Always inspect more than the top file. The ranked SDF and PDB outputs let alternative poses be compared, and a cluster of high-confidence poses that agree on the binding mode is more trustworthy than a single high score. For a physics-based cross-check of the top complex, an MM-PBSA free energy estimate on the ranked structure gives an independent read on binding.
FlowDock fits the case where no holo receptor exists and the pocket may rearrange on binding. It generates the bound protein and ligand together and hands back an affinity estimate in the same run, which classical docking does not.
If a high-quality receptor structure is already in hand and the goal is fast, well-calibrated pose ranking, AutoDock Vina is the leaner choice, and GNINA adds CNN rescoring on the same rigid-receptor setup. For blind docking of a ligand into a known structure without a search box, DiffDock is a closer comparison, though it does not move the protein or score affinity. When the target is a broader complex, including multiple chains or cofactors, the all-atom co-folding models Chai-1, Protenix, and Boltz-2 predict the assembly and, in Boltz-2's case, affinity as well.
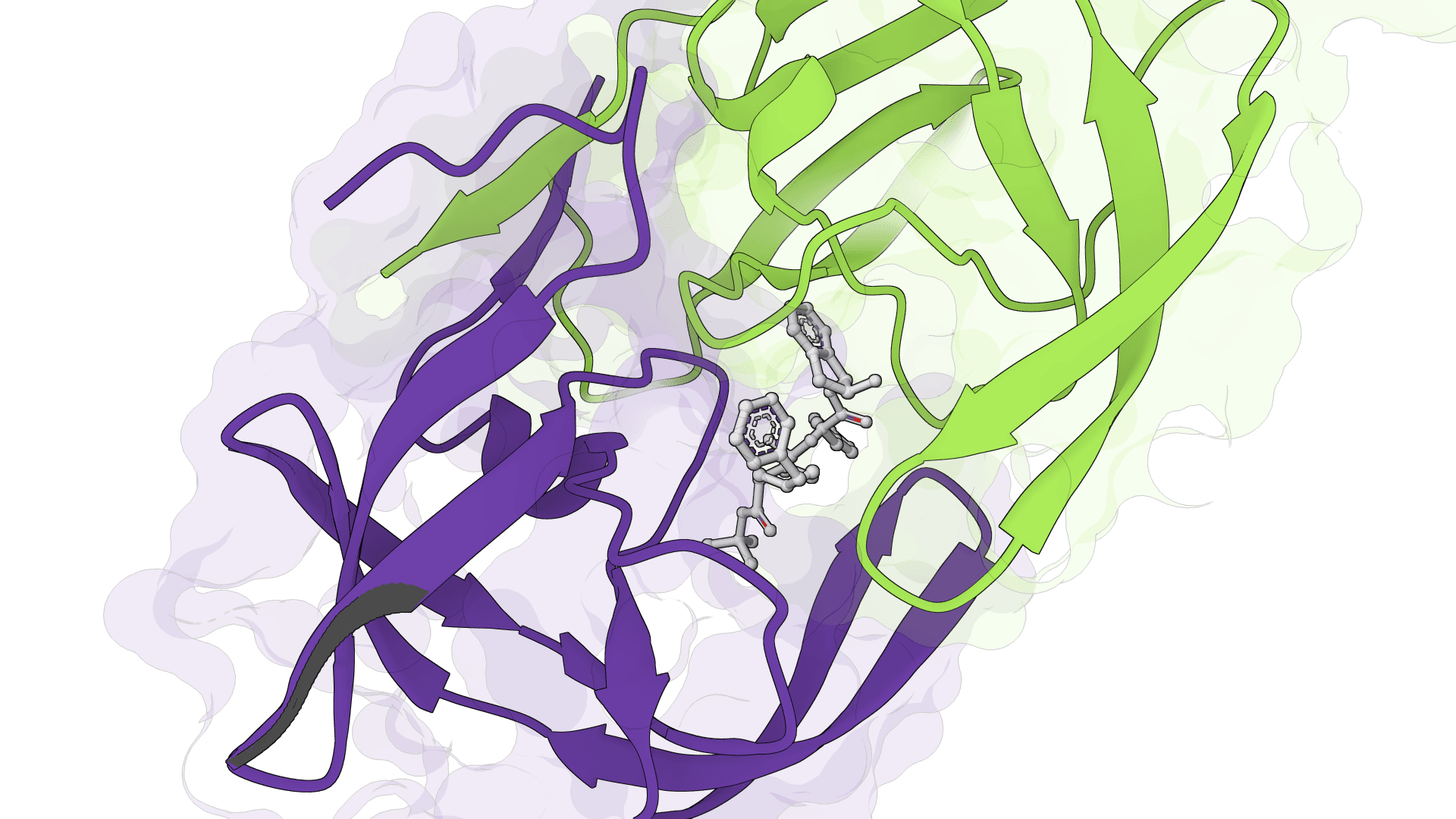
SigmaDock is a fragment-based molecular docking tool using SE(3) equivariant diffusion models to predict how small molecule ligands bind to protein targets. Presented at ICLR 2026, it generates multiple binding poses with Vinardo scoring.
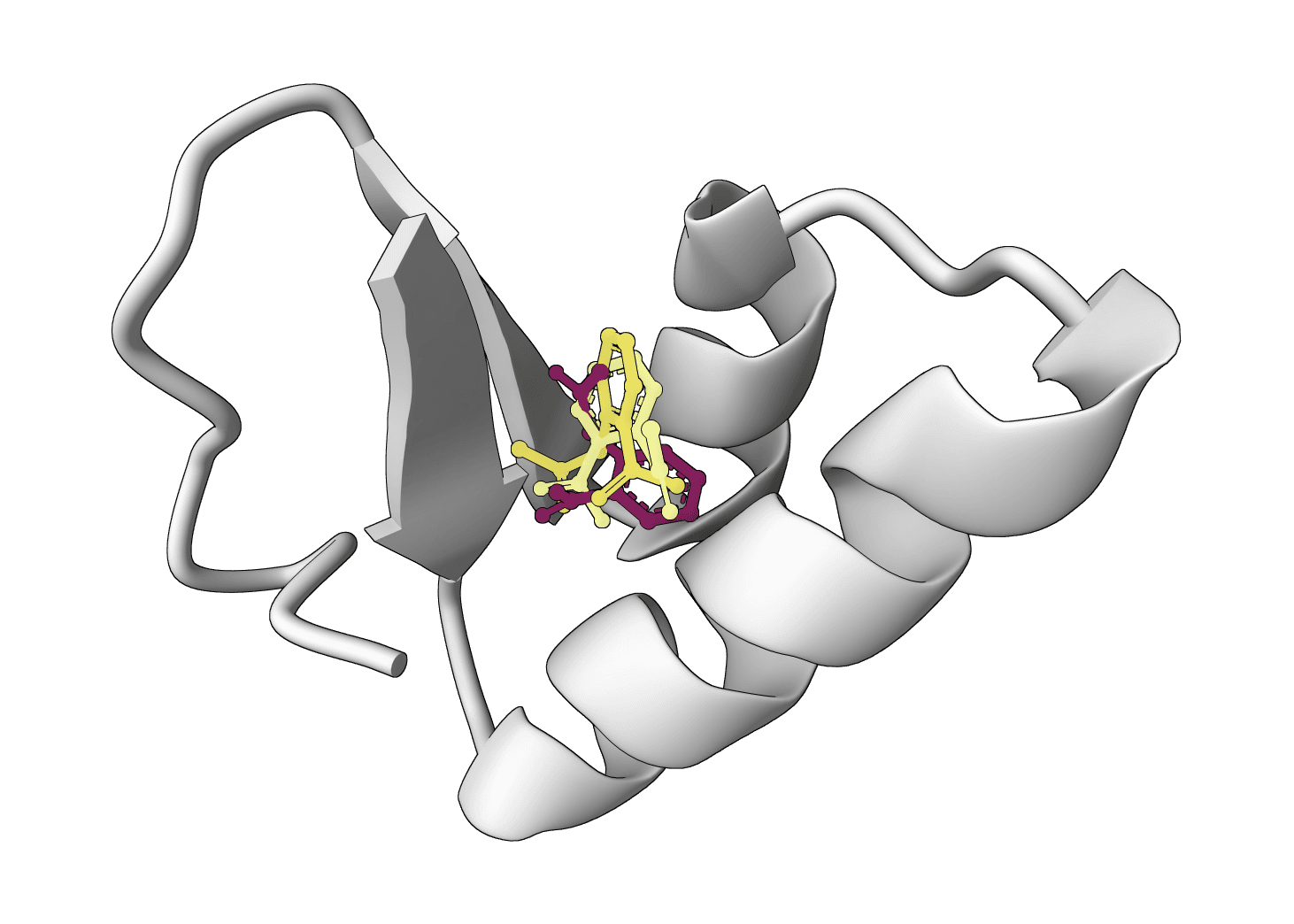
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
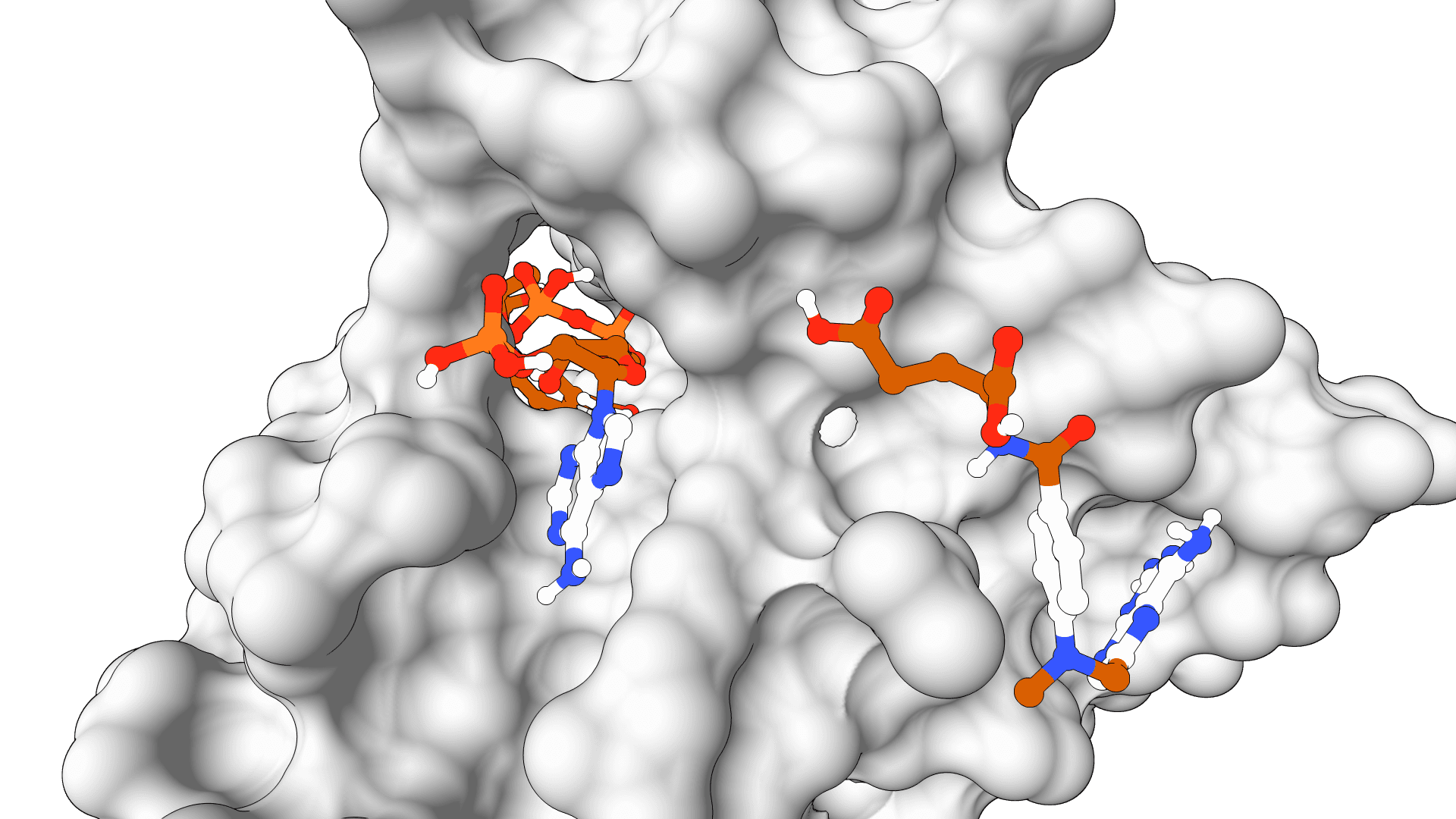
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
AutoDock Vina predicts protein-ligand binding modes with Vina, Vinardo, or AutoDock4 scoring and returns ranked poses with energy estimates.
GNINA is a molecular docking tool that combines traditional physics-based docking with deep learning CNN scoring for protein-small-molecule complexes. It provides accurate binding predictions with confidence scores, optimized for high-throughput virtual screening.
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
TEMPL Pipeline predicts protein-ligand poses by finding similar protein templates, aligning template ligands, generating constrained conformers, and ranking poses with shape and pharmacophore scores.
FlowDock predicts the structure of a protein-ligand complex and estimates how tightly the two bind, starting from nothing more than a protein sequence and a ligand SMILES string. It is a generative model: instead of slotting a ligand into a fixed receptor, it builds the bound protein conformation and the docked ligand pose together, then reports a confidence score and an affinity estimate for each sampled complex.
That single trait separates it from most docking tools. Classic docking assumes a known receptor structure and treats the protein as rigid or nearly so. FlowDock generates the holo (bound) protein from an apo prior, so the backbone and side chains can move to accommodate the ligand. The cost of that flexibility is sampling: FlowDock draws several complexes per run and ranks them, rather than returning a single deterministic answer.
The method comes from Alex Morehead and Jianlin Cheng (ISMB 2025). It is most useful when no experimental holo structure exists, when binding may reshape the pocket, or when a fast affinity estimate alongside the pose is more valuable than a single high-resolution docking result.
ProteinIQ runs FlowDock on GPU infrastructure with no setup. Provide a protein as a sequence, FASTA, or receptor PDB, and a ligand as a SMILES string or SDF file. FlowDock samples several protein-ligand complexes and returns ranked PDB structures, ranked SDF ligand poses, per-pose plDDT confidence, and an affinity estimate for each, all downloadable. An optional template PDB can guide the structure prior.
| Input | Supported formats | Notes |
|---|---|---|
Protein | Plain sequence, FASTA, or PDB | Up to 4096 residues. Separate sequence chains with |. A PDB is used directly as the receptor. |
Ligand | SMILES or SDF | Up to 300 heavy atoms. Use | between SMILES strings for multi-ligand inputs. |
Protein template | PDB (optional) | Used by the ESMFold structure prior when it matches the input sequence. |
Two ProteinIQ tools pair naturally with the inputs here. If only a ligand name or PubChem CID is on hand, fetch the SMILES directly in the ligand card. If the protein is known only by sequence and a starting fold helps, predict one first with ESMFold and pass it as the template.
FlowDock's defaults reproduce the sampling configuration from the original sample.py, so the out-of-the-box run matches the published method. The settings below mostly trade speed against the number and diversity of sampled complexes.
| Setting | Description |
|---|---|
Number of samples | Complexes to generate (1-20, default 5). More samples explore more poses but take longer. Keep it divisible by chunk size. |
Chunk size | Samples processed per batch (1-10, default 5). Number of samples should divide evenly by this value. |
Sampling steps | Integration steps along the flow (10-80, default 40). More steps refine geometry at the cost of runtime. |
Sampler | VDODE (default) or ODE. The variance-diminishing ODE sampler is the published default. |
Sampler eta | Noise scale for the sampler (0.5-2.0, default 1.0). Lower values are more deterministic. |
Start time | 1.0 (default) starts from the full prior; auto lets FlowDock pick the start point. |
Use template | Applies the supplied template PDB to the structure prior when the sequence matches (default on). |
Discard SDF coordinates | Ignores input ligand coordinates and samples poses from scratch (default on). |
Rank by confidence | Orders outputs by the confidence head and writes ranked files (default on). |
Ranking score | Which plDDT drives ranking: Ligand (default), Protein, or Protein + ligand. Ligand plDDT is the right choice when pose quality matters most. |
| Output | Description |
|---|---|
prot_all.pdb | Multi-model PDB with every sampled protein structure. |
lig_all.sdf | Multi-conformer SDF with every sampled ligand pose. |
prot_rank*.pdb | Ranked protein structures (when confidence ranking is on). |
lig_rank*.sdf | Ranked ligand poses (when confidence ranking is on). |
lig_ref.sdf | Reference ligand SDF, written when ligand coordinates are available. |
*_auxiliary_estimation.csv | Per-pose rank, plDDT values, and affinity estimates. |
The Data tab summarizes the same results as a sortable table of rank, plDDT, affinity, and source filename, while the Viewer tab loads the sampled complexes for inspection.
FlowDock treats docking as a transport problem. It learns a continuous flow that carries an unbound starting structure toward a bound protein-ligand complex, parameterized by conditional flow matching. At inference it integrates that flow over a fixed number of steps, moving atoms from the prior toward a plausible holo state.
The starting point comes from a structure prior. FlowDock uses an ESMFold-derived apo conformation of the protein, which is why a sequence is enough to start and why a matching template PDB can be supplied to steer the prior. The ligand is initialized from its molecular graph, with input coordinates discarded by default so the pose is sampled rather than copied.
Two auxiliary heads run on top of the generated geometry. A confidence head produces plDDT-style per-residue and per-ligand scores, and an affinity head predicts binding strength. Because generation is stochastic, FlowDock draws several complexes and uses the confidence head to rank them, so the top-ranked file is the model's best guess rather than the only one it considered.
plDDT here mirrors the confidence metric from structure prediction: higher means the model is more certain about the local geometry it produced. Ligand plDDT speaks to pose reliability, protein plDDT to the backbone and pocket. Treat a high-plDDT top pose as a confident prediction and a run where even the best pose scores low as a signal that the pocket, the ligand, or the prior is hard for the model.
The affinity values are model estimates, not measurements. Use them to rank candidates against each other within a campaign, not as a substitute for an assay or a physics-based free energy. When ranking matters, compare relative affinity across ligands docked the same way rather than reading any single number as an absolute Kd.
Always inspect more than the top file. The ranked SDF and PDB outputs let alternative poses be compared, and a cluster of high-confidence poses that agree on the binding mode is more trustworthy than a single high score. For a physics-based cross-check of the top complex, an MM-PBSA free energy estimate on the ranked structure gives an independent read on binding.
FlowDock fits the case where no holo receptor exists and the pocket may rearrange on binding. It generates the bound protein and ligand together and hands back an affinity estimate in the same run, which classical docking does not.
If a high-quality receptor structure is already in hand and the goal is fast, well-calibrated pose ranking, AutoDock Vina is the leaner choice, and GNINA adds CNN rescoring on the same rigid-receptor setup. For blind docking of a ligand into a known structure without a search box, DiffDock is a closer comparison, though it does not move the protein or score affinity. When the target is a broader complex, including multiple chains or cofactors, the all-atom co-folding models Chai-1, Protenix, and Boltz-2 predict the assembly and, in Boltz-2's case, affinity as well.
SigmaDock is a fragment-based molecular docking tool using SE(3) equivariant diffusion models to predict how small molecule ligands bind to protein targets. Presented at ICLR 2026, it generates multiple binding poses with Vinardo scoring.
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
AutoDock Vina predicts protein-ligand binding modes with Vina, Vinardo, or AutoDock4 scoring and returns ranked poses with energy estimates.
GNINA is a molecular docking tool that combines traditional physics-based docking with deep learning CNN scoring for protein-small-molecule complexes. It provides accurate binding predictions with confidence scores, optimized for high-throughput virtual screening.
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
TEMPL Pipeline predicts protein-ligand poses by finding similar protein templates, aligning template ligands, generating constrained conformers, and ranking poses with shape and pharmacophore scores.