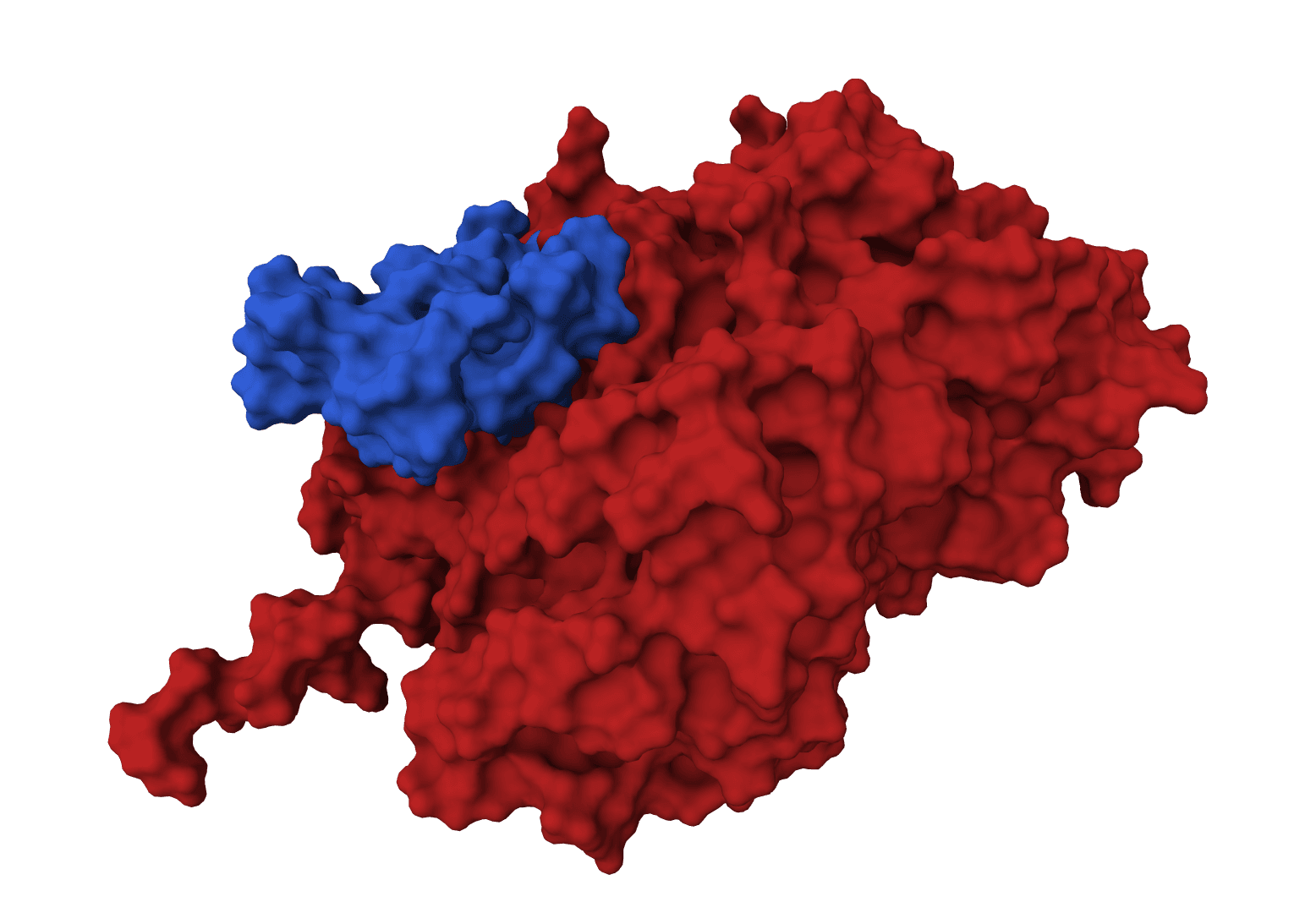
GeoDock predicts how two proteins assemble into a complex. Given two separate structures, it places them together and lets each backbone and side chain shift to fit its partner, so the result reflects the conformational change that happens on binding rather than a rigid lock-and-key fit.
That flexibility is the point. Classical docking treats both partners as solid bodies and samples millions of orientations, which fails when binding reshapes a loop or repacks an interface. GeoDock instead learns the geometry of real complexes from the DIPS dataset and predicts the bound arrangement directly in a single pass that runs in about a second on a GPU.
Upload two protein structures on ProteinIQ, one as the receptor and one as the ligand partner, then run the DIPS 0.3 model to get a predicted complex back. Each partner can be a PDB file or an RCSB PDB ID. The output is a docked complex in PDB format with per-residue pLDDT confidence written into the B-factor column, viewable in 3D and downloadable.
Both partners are protein structures. GeoDock docks them as given, so the input conformations matter: feed it the unbound or predicted monomer structures you actually want to assemble.
| Input | Description |
|---|---|
Receptor (first protein partner) | PDB file (.pdb or .ent) or RCSB PDB ID. Up to 1000 residues. |
Ligand (second protein partner) | PDB file (.pdb or .ent) or RCSB PDB ID. Up to 1000 residues. |
The combined size of both partners is capped at 1200 residues to stay within GPU memory. The naming follows docking convention: receptor and ligand here are both proteins, not a small-molecule ligand. For small-molecule docking, use GNINA instead.
If you only have sequences, predict each monomer first with ESMFold or AlphaFold 2, then dock the resulting structures.
| Setting | Description |
|---|---|
OpenMM refinement | On by default. Relaxes the predicted complex with OpenMM energy minimization (amber14 ff14SB) to remove steric clashes and add hydrogens. Turn off to return the raw model output. |
Refinement cleans up the interface but does not move the partners far from where the model placed them. Turning it off keeps the prediction exactly as the network produced it, which is useful when comparing raw model geometry across runs.
| Column | Meaning |
|---|---|
Rank | Order of the returned complex. |
Mean pLDDT | Average per-residue confidence across the complex (0-100). |
Min pLDDT | Lowest per-residue confidence, often at flexible termini or loops. |
Max pLDDT | Highest per-residue confidence, usually in well-defined cores. |
File | The complex PDB file name. |
GeoDock reuses the pLDDT scale familiar from AlphaFold and writes a per-residue value into the B-factor column of the output PDB, so any viewer can color the model by confidence.
| pLDDT | Meaning |
|---|---|
| > 90 | High confidence, residue position likely accurate |
| 70-90 | Reasonable, backbone probably correct |
| 50-70 | Low confidence, treat with caution |
| < 50 | Unreliable, may be flexible or disordered |
Confidence concentrates at the interface for a well-predicted complex. Low pLDDT spread across the binding region is a signal the docked pose is uncertain, not just that a terminus is floppy. Because GeoDock returns a single predicted arrangement rather than a scored ensemble, the pLDDT pattern at the interface is the main internal guide to how much to trust the pose.
GeoDock is a multi-track iterative transformer. It carries two representations in parallel, a per-residue track and a pairwise track that encodes relative geometry between residues across both partners, and refines them over repeated passes. From the final pairwise representation it predicts the rigid-body transform that brings the partners together along with the residue-level adjustments that flex each backbone into its bound state.
The model was trained on the DIPS dataset of experimentally determined complexes, and the served checkpoint is DIPS 0.3 from the Gray Lab release. Inference is direct: there is no global search over orientations and no scoring of many candidate poses, which is why a prediction finishes in roughly a second per pair.
GeoDock fits when both binding partners are known proteins and you want a fast, flexible prediction of how they come together. It is strongest as a quick first model of a binary complex, especially for screening many candidate pairs where running a slower method on each is impractical.
For protein-protein docking with a diffusion-based generative approach that samples multiple poses, compare against DFMDock. For docking a small molecule into a protein rather than two proteins together, use GNINA. When a binding interface is reshaped dramatically or many copies assemble into a larger oligomer, a full co-folding model handles those cases better than pairwise docking.
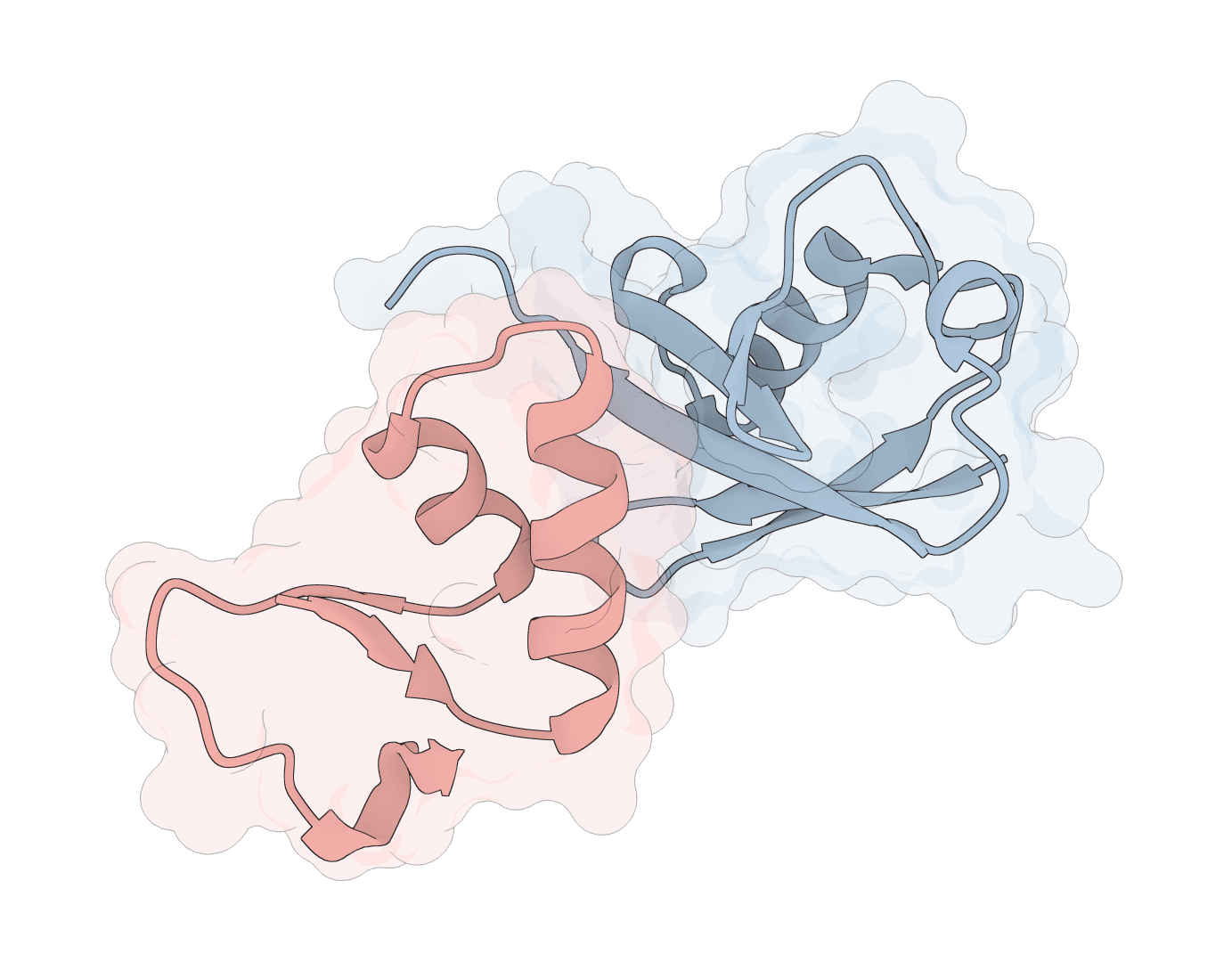
AF2Dock adapts AlphaFold2-style co-folding for structure-based protein-protein docking. It docks receptor and ligand protein structures with flow-matching refinement and ranks sampled complexes by iPTM.
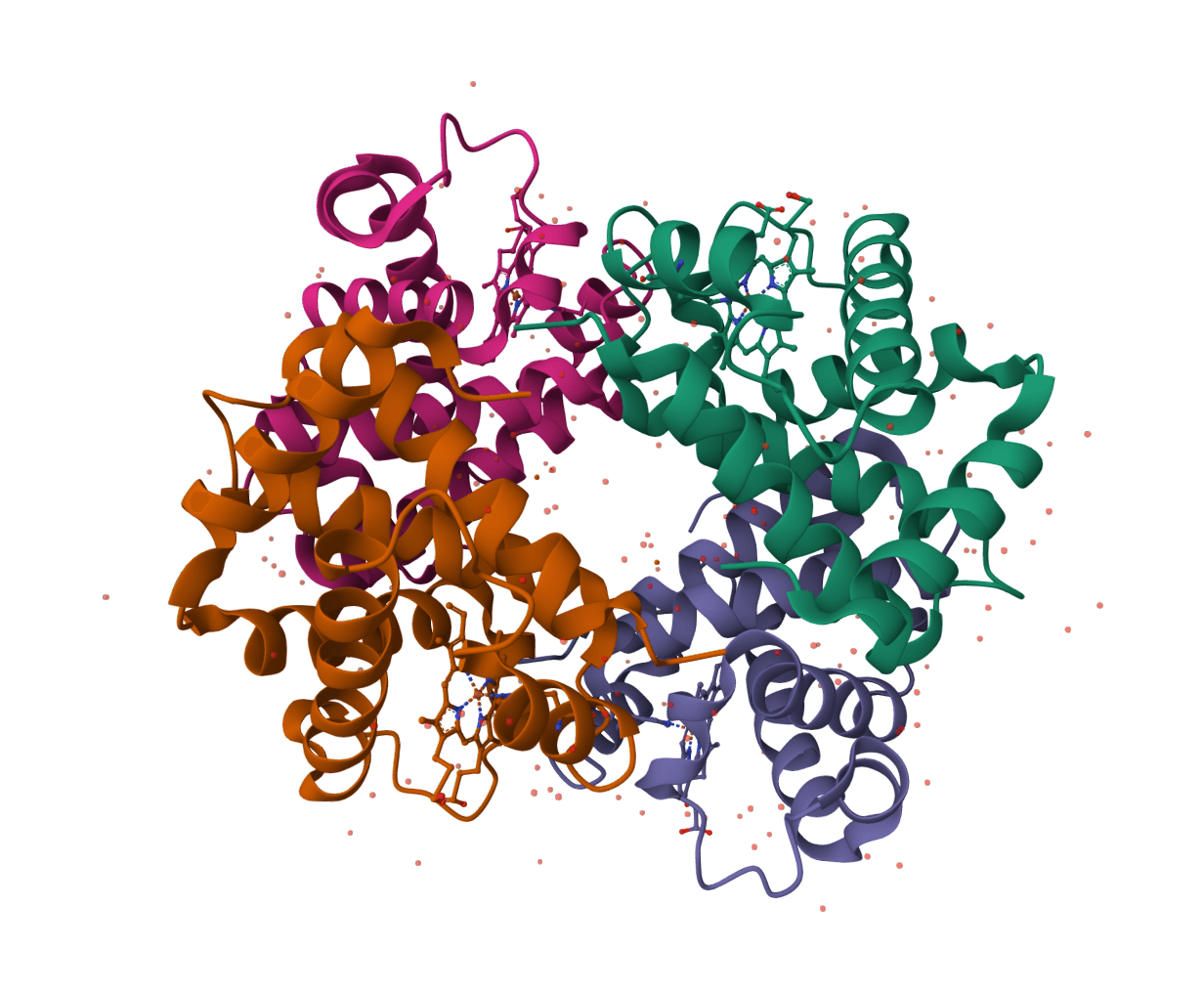
DFMDock (Denoising Force Matching Dock) is a diffusion model that unifies sampling and ranking for protein-protein docking within a single framework. It predicts docked poses for protein-protein complexes from unbound structures using denoising score matching with optional clash force guidance.
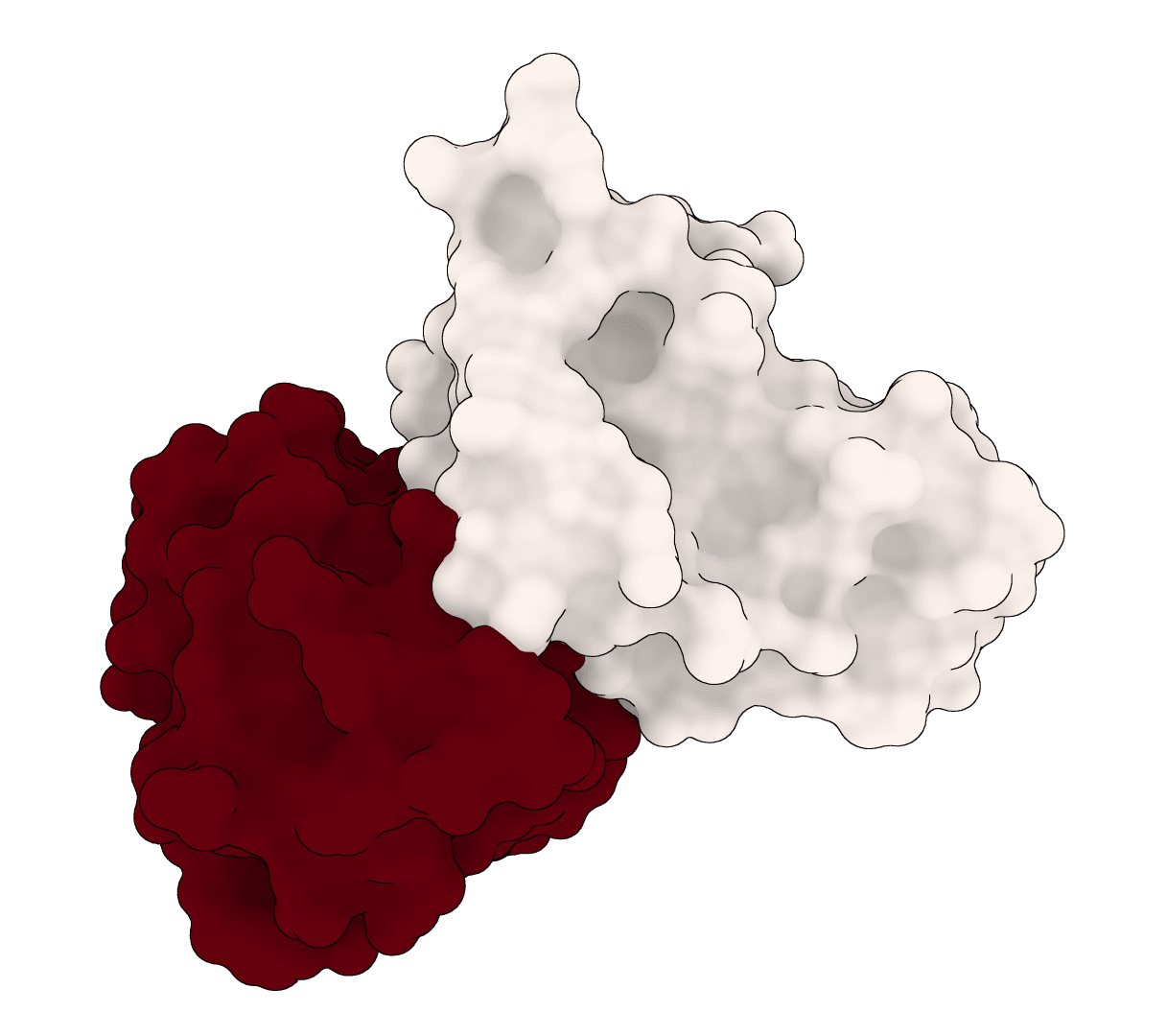
EquiDock is an SE(3)-equivariant graph neural network for rigid protein-protein docking. It predicts a binding pose for a protein-protein complex from unbound structures using geometric deep learning, with DIPS and DB5 pretrained checkpoints from the native release.
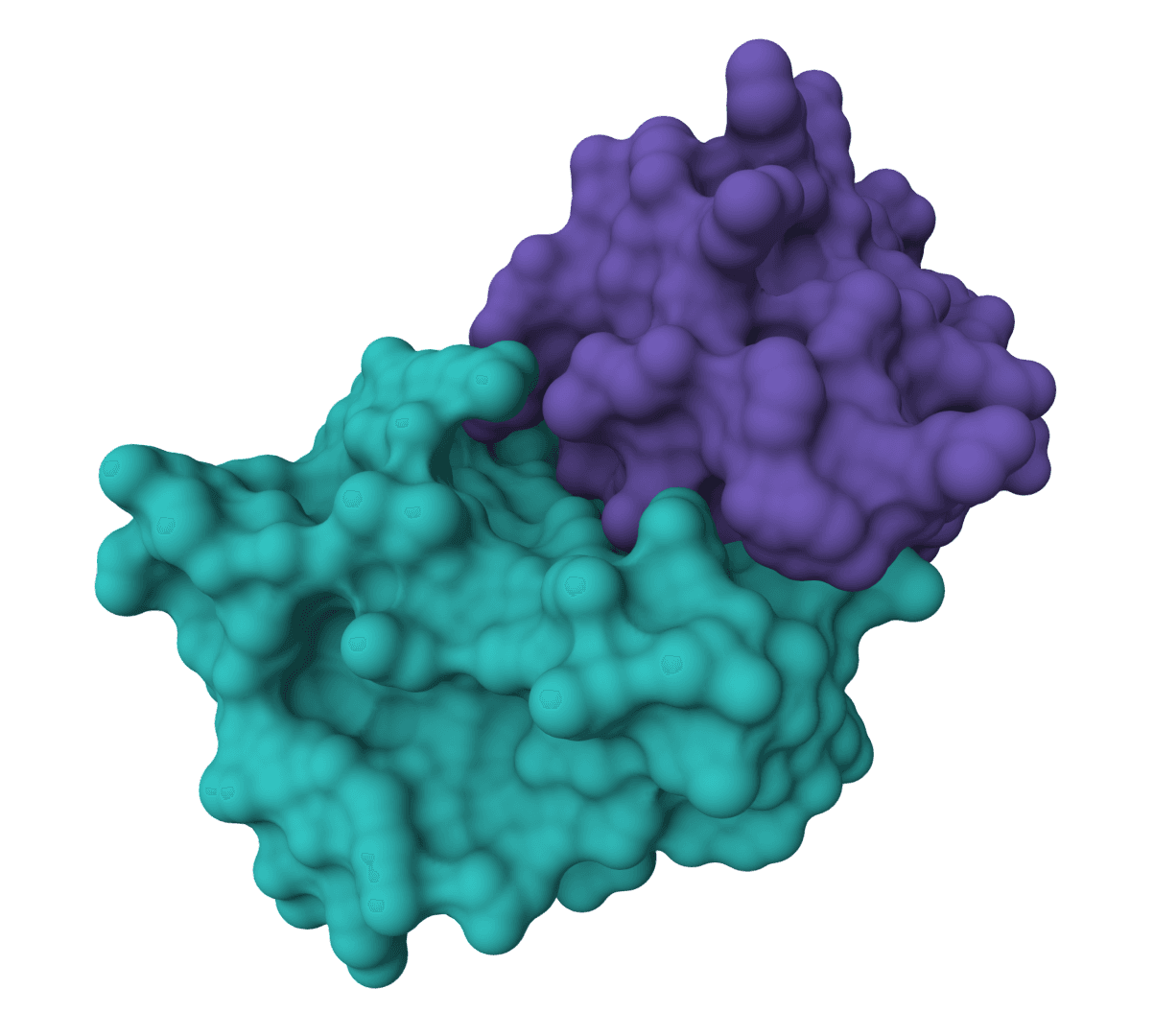
ColabDock is a protein-protein docking framework that uses AlphaFold2 to predict complex structures guided by experimental restraints from cross-linking mass spectrometry, NMR, or other sources.
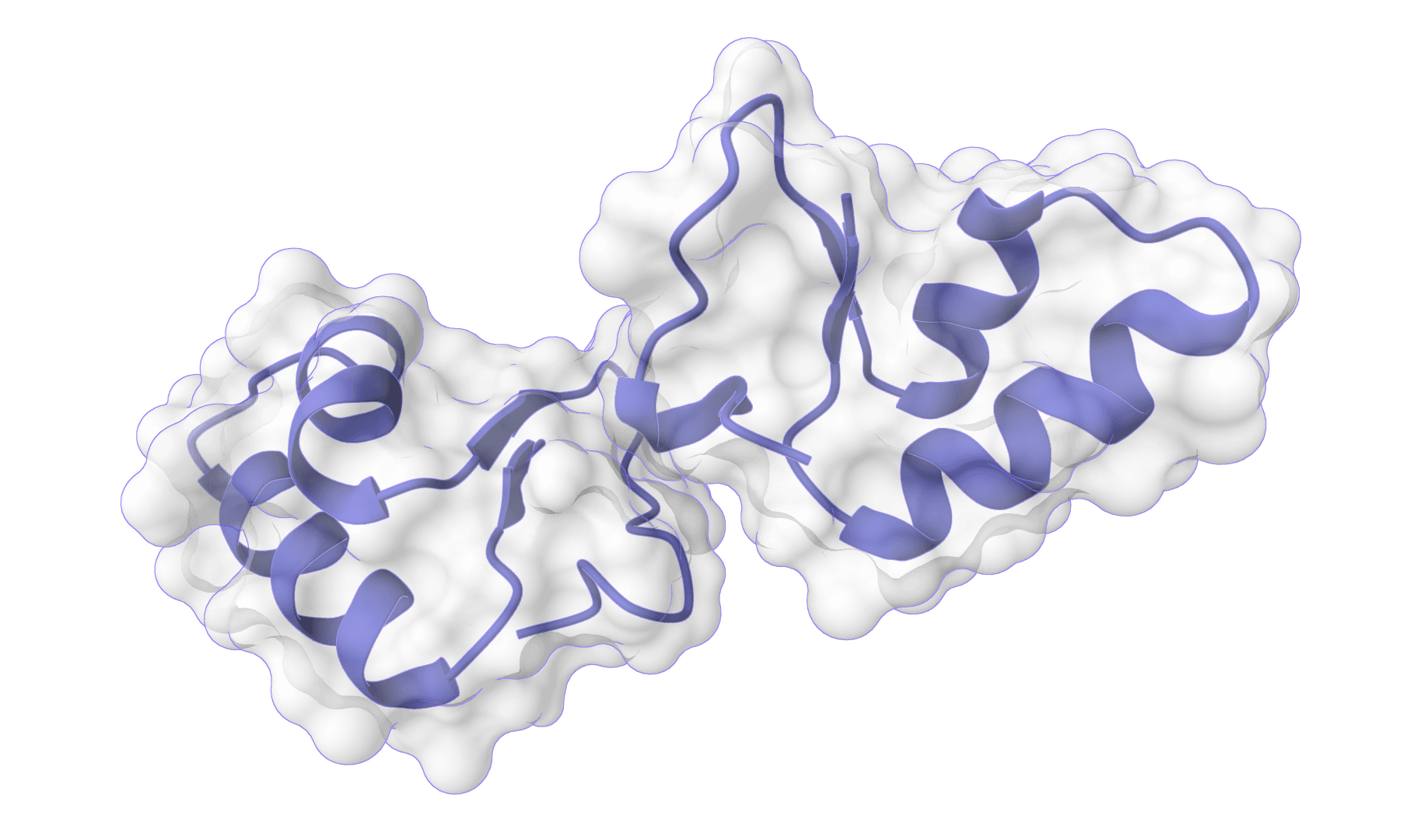
HADDOCK (High Ambiguity Driven protein-protein DOCKing) is an integrative modeling platform for biomolecular complexes. It uses experimental data and bioinformatic predictions to guide the docking process, generating accurate protein-protein complex structures.
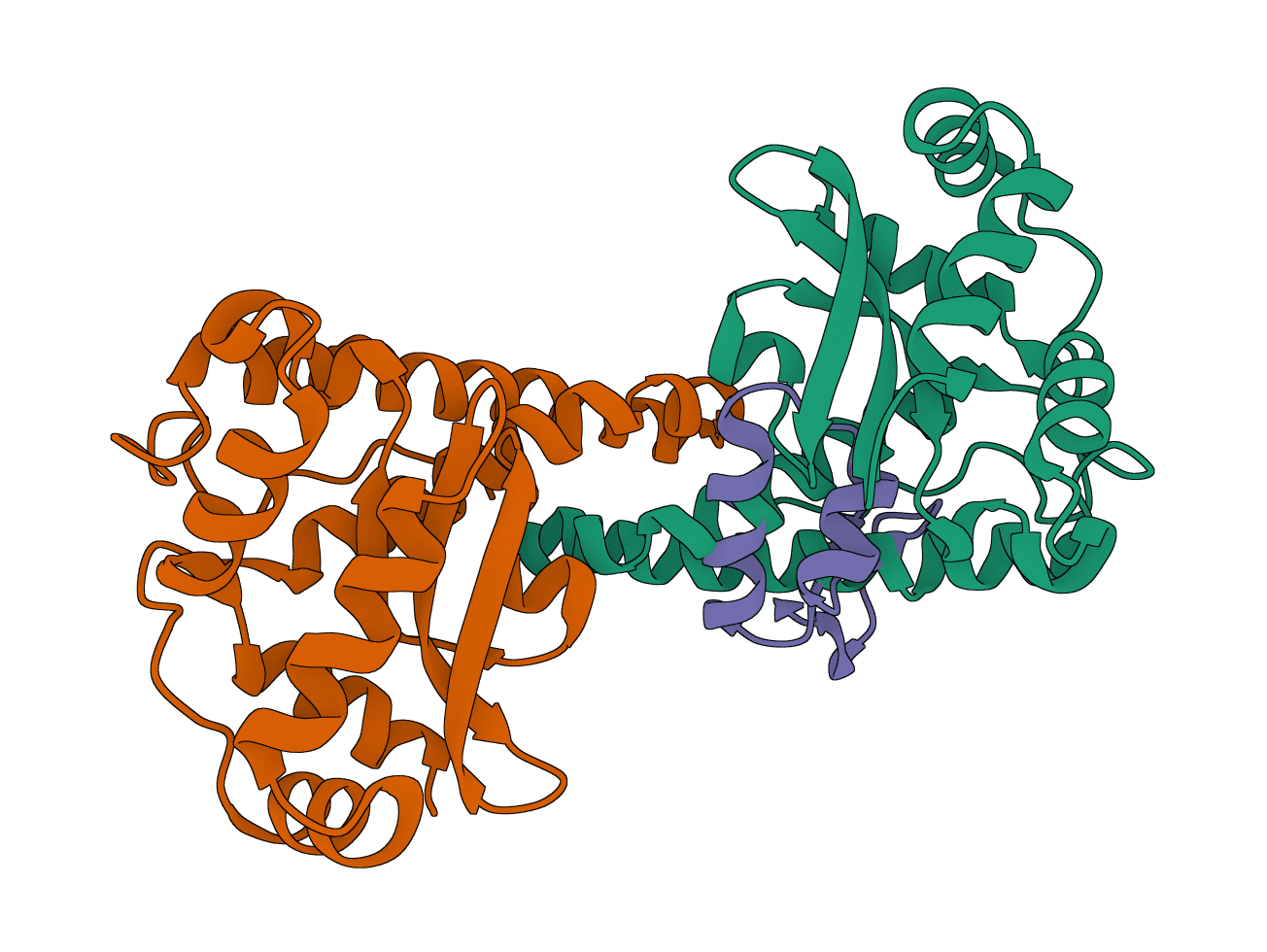
LightDock is a protein-protein, protein-peptide, and protein-DNA docking framework using Glowworm Swarm Optimization (GSO). It predicts macromolecular binding modes and interfaces for biological complexes.
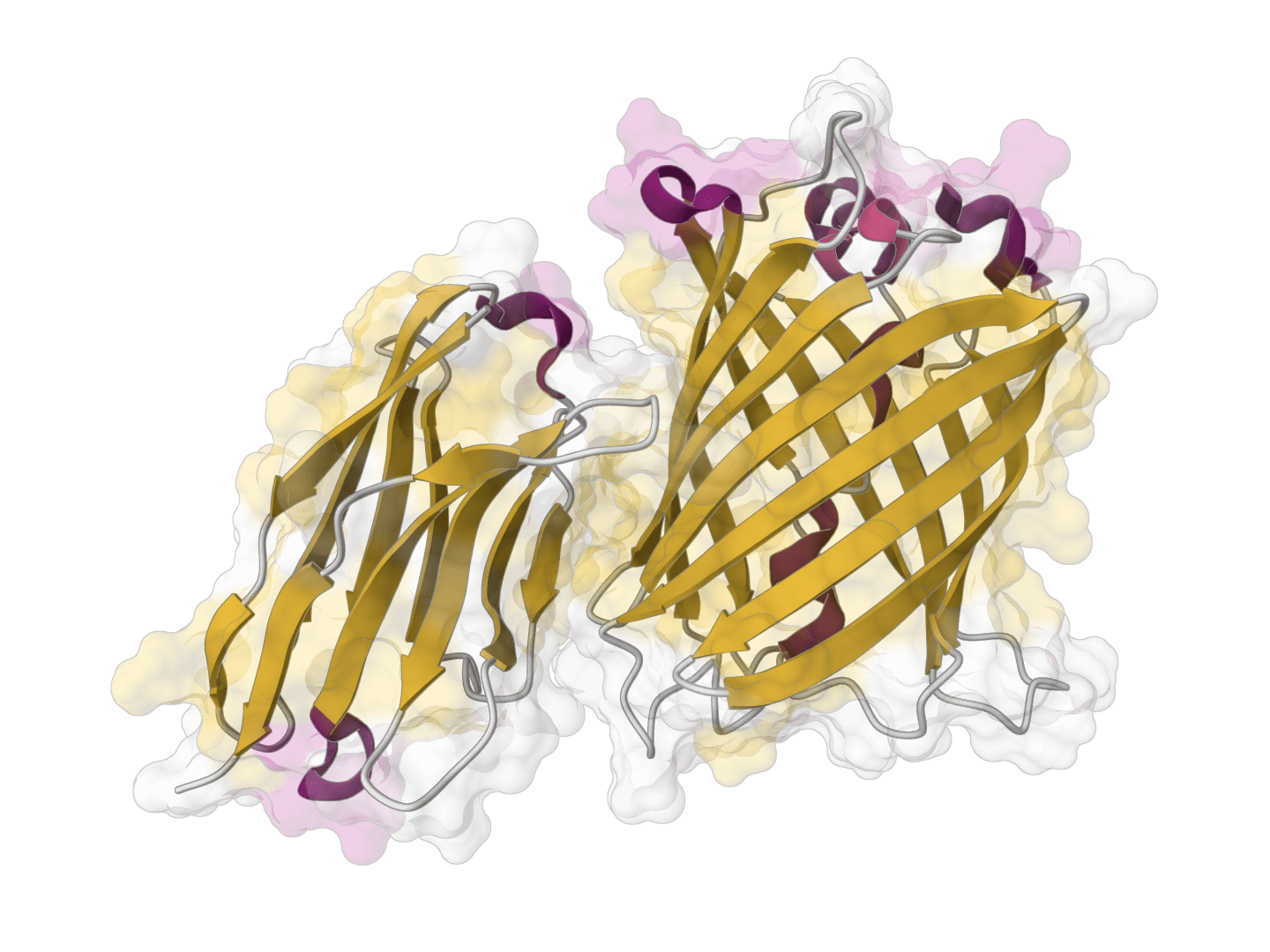
ParaSurf is a state-of-the-art surface-based deep learning model for predicting interactions between antibodies and antigens. It identifies paratope binding sites on antibody structures with high accuracy across multiple benchmark datasets.
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
GeoDock predicts how two proteins assemble into a complex. Given two separate structures, it places them together and lets each backbone and side chain shift to fit its partner, so the result reflects the conformational change that happens on binding rather than a rigid lock-and-key fit.
That flexibility is the point. Classical docking treats both partners as solid bodies and samples millions of orientations, which fails when binding reshapes a loop or repacks an interface. GeoDock instead learns the geometry of real complexes from the DIPS dataset and predicts the bound arrangement directly in a single pass that runs in about a second on a GPU.
Upload two protein structures on ProteinIQ, one as the receptor and one as the ligand partner, then run the DIPS 0.3 model to get a predicted complex back. Each partner can be a PDB file or an RCSB PDB ID. The output is a docked complex in PDB format with per-residue pLDDT confidence written into the B-factor column, viewable in 3D and downloadable.
Both partners are protein structures. GeoDock docks them as given, so the input conformations matter: feed it the unbound or predicted monomer structures you actually want to assemble.
| Input | Description |
|---|---|
Receptor (first protein partner) | PDB file (.pdb or .ent) or RCSB PDB ID. Up to 1000 residues. |
Ligand (second protein partner) | PDB file (.pdb or .ent) or RCSB PDB ID. Up to 1000 residues. |
The combined size of both partners is capped at 1200 residues to stay within GPU memory. The naming follows docking convention: receptor and ligand here are both proteins, not a small-molecule ligand. For small-molecule docking, use GNINA instead.
If you only have sequences, predict each monomer first with ESMFold or AlphaFold 2, then dock the resulting structures.
| Setting | Description |
|---|---|
OpenMM refinement | On by default. Relaxes the predicted complex with OpenMM energy minimization (amber14 ff14SB) to remove steric clashes and add hydrogens. Turn off to return the raw model output. |
Refinement cleans up the interface but does not move the partners far from where the model placed them. Turning it off keeps the prediction exactly as the network produced it, which is useful when comparing raw model geometry across runs.
| Column | Meaning |
|---|---|
Rank | Order of the returned complex. |
Mean pLDDT | Average per-residue confidence across the complex (0-100). |
Min pLDDT | Lowest per-residue confidence, often at flexible termini or loops. |
Max pLDDT | Highest per-residue confidence, usually in well-defined cores. |
File | The complex PDB file name. |
GeoDock reuses the pLDDT scale familiar from AlphaFold and writes a per-residue value into the B-factor column of the output PDB, so any viewer can color the model by confidence.
| pLDDT | Meaning |
|---|---|
| > 90 | High confidence, residue position likely accurate |
| 70-90 | Reasonable, backbone probably correct |
| 50-70 | Low confidence, treat with caution |
| < 50 | Unreliable, may be flexible or disordered |
Confidence concentrates at the interface for a well-predicted complex. Low pLDDT spread across the binding region is a signal the docked pose is uncertain, not just that a terminus is floppy. Because GeoDock returns a single predicted arrangement rather than a scored ensemble, the pLDDT pattern at the interface is the main internal guide to how much to trust the pose.
GeoDock is a multi-track iterative transformer. It carries two representations in parallel, a per-residue track and a pairwise track that encodes relative geometry between residues across both partners, and refines them over repeated passes. From the final pairwise representation it predicts the rigid-body transform that brings the partners together along with the residue-level adjustments that flex each backbone into its bound state.
The model was trained on the DIPS dataset of experimentally determined complexes, and the served checkpoint is DIPS 0.3 from the Gray Lab release. Inference is direct: there is no global search over orientations and no scoring of many candidate poses, which is why a prediction finishes in roughly a second per pair.
GeoDock fits when both binding partners are known proteins and you want a fast, flexible prediction of how they come together. It is strongest as a quick first model of a binary complex, especially for screening many candidate pairs where running a slower method on each is impractical.
For protein-protein docking with a diffusion-based generative approach that samples multiple poses, compare against DFMDock. For docking a small molecule into a protein rather than two proteins together, use GNINA. When a binding interface is reshaped dramatically or many copies assemble into a larger oligomer, a full co-folding model handles those cases better than pairwise docking.
AF2Dock adapts AlphaFold2-style co-folding for structure-based protein-protein docking. It docks receptor and ligand protein structures with flow-matching refinement and ranks sampled complexes by iPTM.
DFMDock (Denoising Force Matching Dock) is a diffusion model that unifies sampling and ranking for protein-protein docking within a single framework. It predicts docked poses for protein-protein complexes from unbound structures using denoising score matching with optional clash force guidance.
EquiDock is an SE(3)-equivariant graph neural network for rigid protein-protein docking. It predicts a binding pose for a protein-protein complex from unbound structures using geometric deep learning, with DIPS and DB5 pretrained checkpoints from the native release.
ColabDock is a protein-protein docking framework that uses AlphaFold2 to predict complex structures guided by experimental restraints from cross-linking mass spectrometry, NMR, or other sources.
HADDOCK (High Ambiguity Driven protein-protein DOCKing) is an integrative modeling platform for biomolecular complexes. It uses experimental data and bioinformatic predictions to guide the docking process, generating accurate protein-protein complex structures.
LightDock is a protein-protein, protein-peptide, and protein-DNA docking framework using Glowworm Swarm Optimization (GSO). It predicts macromolecular binding modes and interfaces for biological complexes.
ParaSurf is a state-of-the-art surface-based deep learning model for predicting interactions between antibodies and antigens. It identifies paratope binding sites on antibody structures with high accuracy across multiple benchmark datasets.
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.