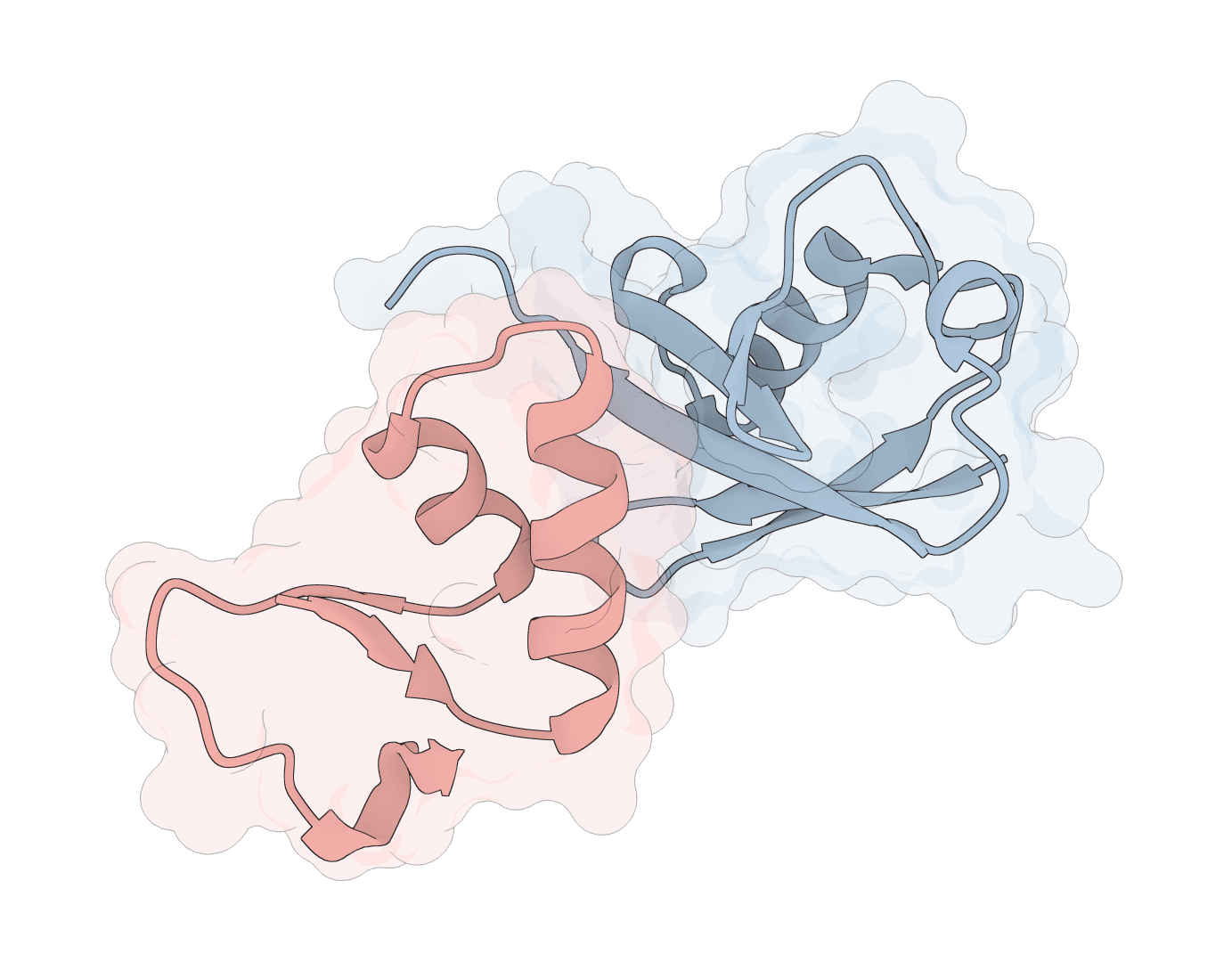
AF2Dock is a Gray Lab method for structure-based protein-protein docking. It adapts AlphaFold2-style co-folding to start from two protein structures, then refines receptor-ligand placement through flow matching and ranks sampled complexes with iPTM.
Use AF2Dock when you already have receptor and ligand structures and want a set of ranked complex models without manually defining interface restraints. The default hosted model uses the AF2Dock_base checkpoint.
Upload a receptor protein structure and a ligand protein structure, or fetch each partner from RCSB. AF2Dock accepts .pdb and .cif files. RCSB entries are fetched as CIF structures by default because AF2Dock checks file suffixes directly and supports pdb and cif.
If either structure has unresolved residues that must be mapped back to a full chain sequence, add the optional A3M alignment for that partner. The A3M must include two records for every chain: for chain A, name them A_full and A. Those records must have the same aligned length, and the non-gap positions in the chain record must equal the resolved residue count in the uploaded structure.
| Input | Accepted format | Description |
|---|---|---|
Receptor structure | .pdb, .cif, or RCSB ID | First protein docking partner. Protein ATOM records are required. |
Ligand structure | .pdb, .cif, or RCSB ID | Second protein docking partner moved during docking. Protein ATOM records are required. |
Receptor A3M alignment | .a3m | Optional mapping from resolved receptor residues to full receptor sequence when residues are missing. |
Ligand A3M alignment | .a3m | Optional mapping from resolved ligand residues to full ligand sequence when residues are missing. |
| Setting | Default | Description |
|---|---|---|
Number of samples | 1 | Number of sampled complexes to generate. Increase toward the native 40-pose source setting when you need alternative interfaces. |
Flow-matching steps | 10 | Native --num_steps value. More steps increase refinement time. |
Merge first steps | 0 | Native --merge_first_n_steps value. Leave at 0 for the standard run. |
Additional refine steps | 0 | Native --additional_refine_steps value. Adds refinement after interpolation. |
Random seed | blank | Blank lets AF2Dock choose a random seed. Enter a non-negative integer for reproducibility. |
Filter low-pLDDT residues | false | Leave off to preserve the default behavior of no input pLDDT cutoff. |
ProteinIQ caps hosted jobs at 40 samples, 50 flow-matching steps, 2,200 total residues, and 62 combined receptor-ligand chains so jobs fit the shared GPU runtime and AF2Dock's PDB output format.
AF2Dock writes a target folder for each submitted receptor-ligand pair. ProteinIQ returns every generated file from that folder.
| File pattern | Description |
|---|---|
*_iptm.csv | Ranked sample table sorted by iPTM from high to low. |
*_sN.pdb | Final predicted complex for sample N, with pLDDT values written into the B-factor column. |
*_sN_ori_chain.pdb | Predicted complex with original residue and chain mapping restored when AF2Dock can recover it. |
*_sN_out.pkl | Confidence and auxiliary output dictionary for sample N, including pLDDT, pTM, iPTM, weighted pTM, PAE, final atom positions, and final atom mask. |
*_sN_t0_template.pdb | Initial template written by AF2Dock at the first step. |
The Metrics tab is sorted by iPTM. Higher iPTM generally indicates a stronger model confidence signal for the sampled interface within the same AF2Dock job. Treat iPTM as a ranking signal for poses from one run, not as an experimental affinity measurement.
Start with the top-ranked complex in the 3D viewer, then compare nearby ranks for alternative interfaces. The *_ori_chain.pdb files are useful when you need to trace residues back to original chain IDs, while the *_out.pkl files preserve detailed confidence arrays for offline analysis.
The first-step *_t0_template.pdb file is returned because AF2Dock writes it during inference even when full intermediate output saving is disabled. If you enable intermediate prediction or confidence settings, additional step-level PDB or pickle files may be included.
| Tool | Best fit | Why choose it |
|---|---|---|
AF2Dock | Protein-protein docking from two structures with AF2-style refinement | Returns ranked complex structures and AF2-style confidence outputs. |
| DFMDock | Fast rigid docking without MSAs or AlphaFold-style templates | Useful for exploratory structure-only docking with learned energy ranking. |
| HADDOCK3 | Experimental restraints or interface residues are available | Integrates explicit docking restraints and refinement stages. |
| ColabDock | AlphaFold2-guided docking with manual or reference-derived restraints | Better fit when restraint information should steer complex prediction. |
| DockQ | Evaluate a predicted complex against a known reference | Use after docking when a native or benchmark complex is available. |
| PPAP | Estimate protein-protein affinity for a predicted complex | Use after docking when ranking or triaging protein interfaces by affinity is needed. |
AF2Dock is most useful when receptor and ligand structures are available and the docking question benefits from AlphaFold2-style confidence outputs. It is less suitable for non-protein ligands, nucleic-acid partners, cofactors, large induced-fit motions, or structures whose missing residues cannot be mapped with an A3M file.
Based on Graylab/AF2Dock v1.0.0 and the AF2Dock_base model checkpoint.
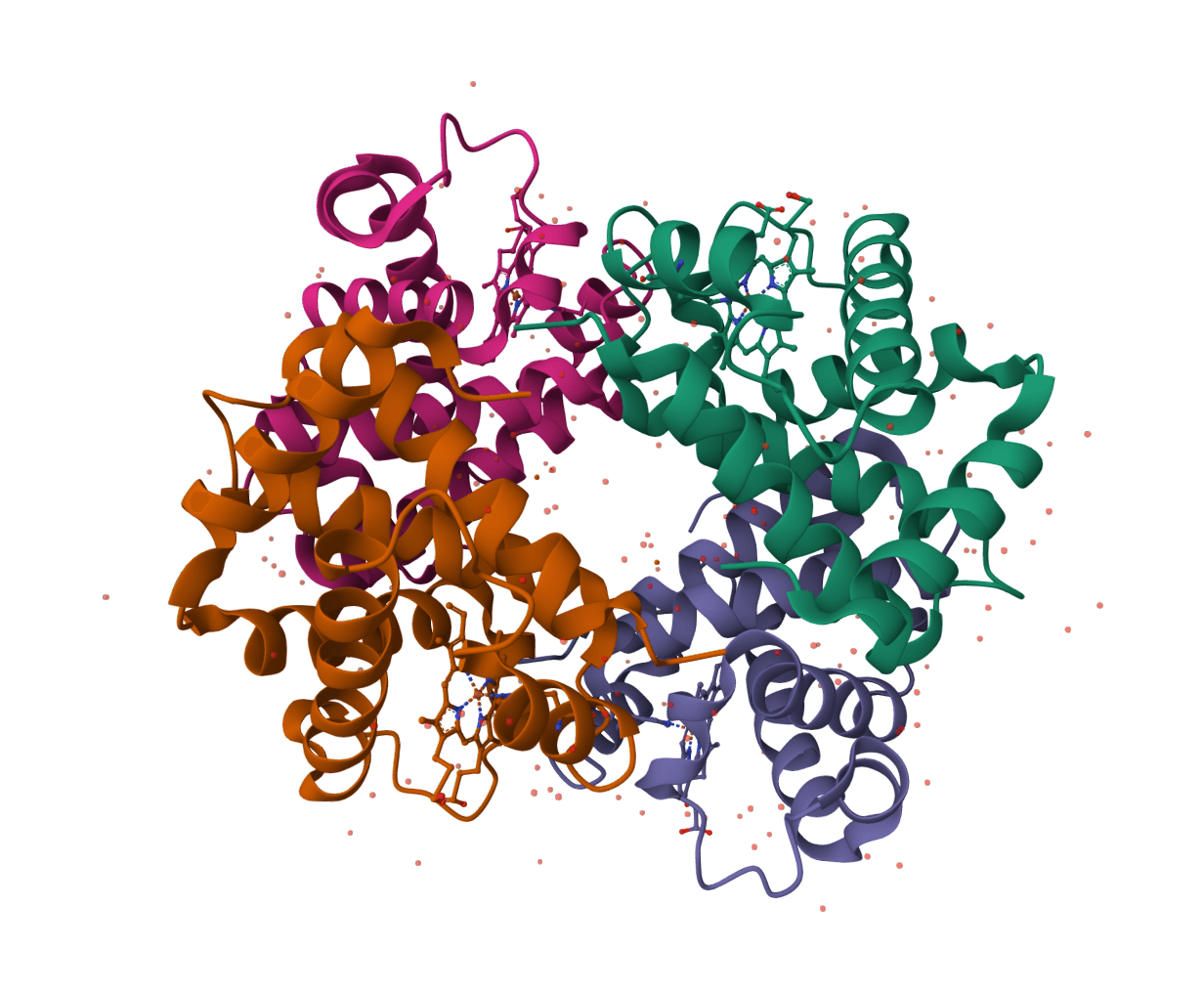
DFMDock (Denoising Force Matching Dock) is a diffusion model that unifies sampling and ranking for protein-protein docking within a single framework. It predicts docked poses for protein-protein complexes from unbound structures using denoising score matching with optional clash force guidance.
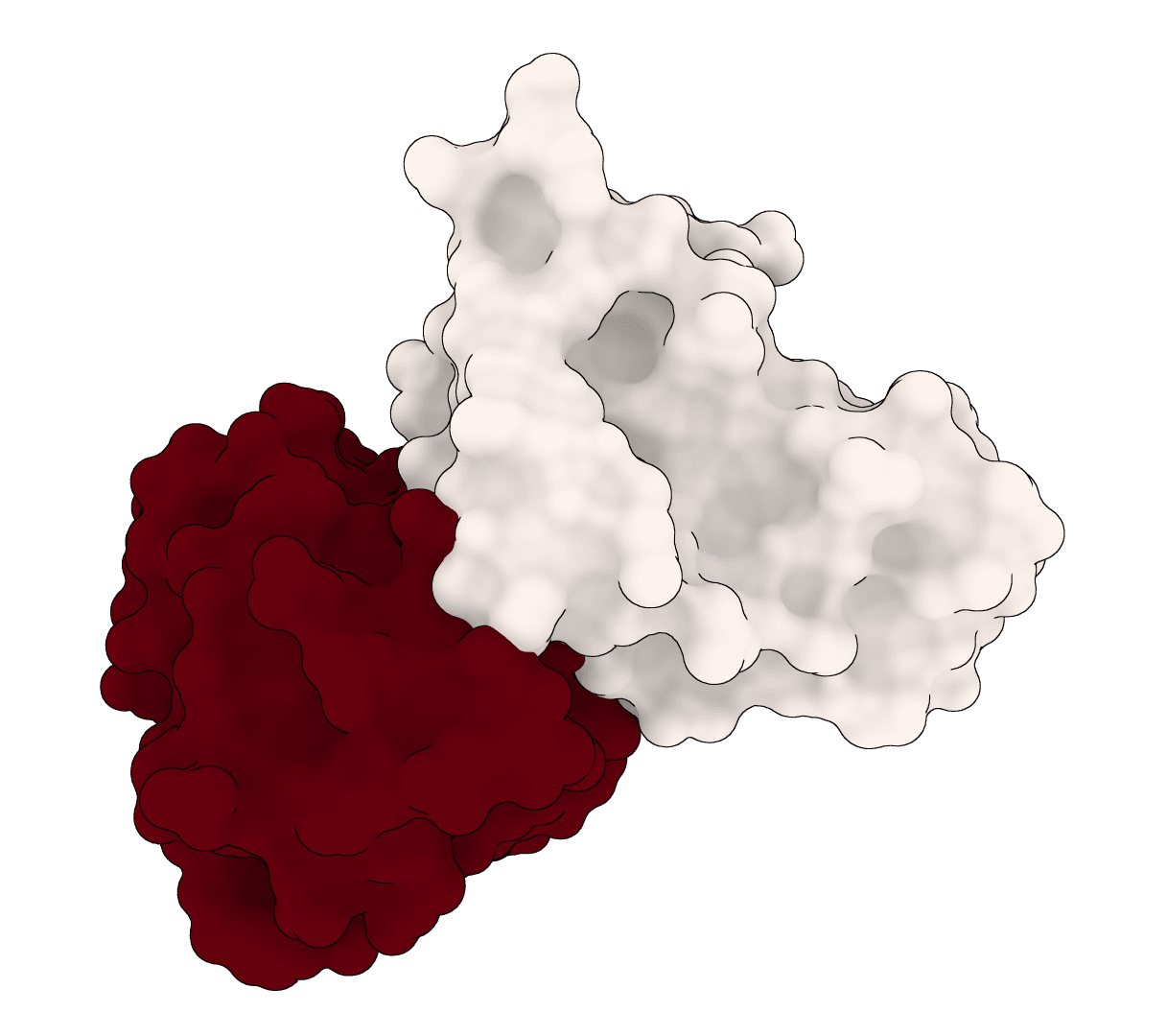
EquiDock is an SE(3)-equivariant graph neural network for rigid protein-protein docking. It predicts a binding pose for a protein-protein complex from unbound structures using geometric deep learning, with DIPS and DB5 pretrained checkpoints from the native release.
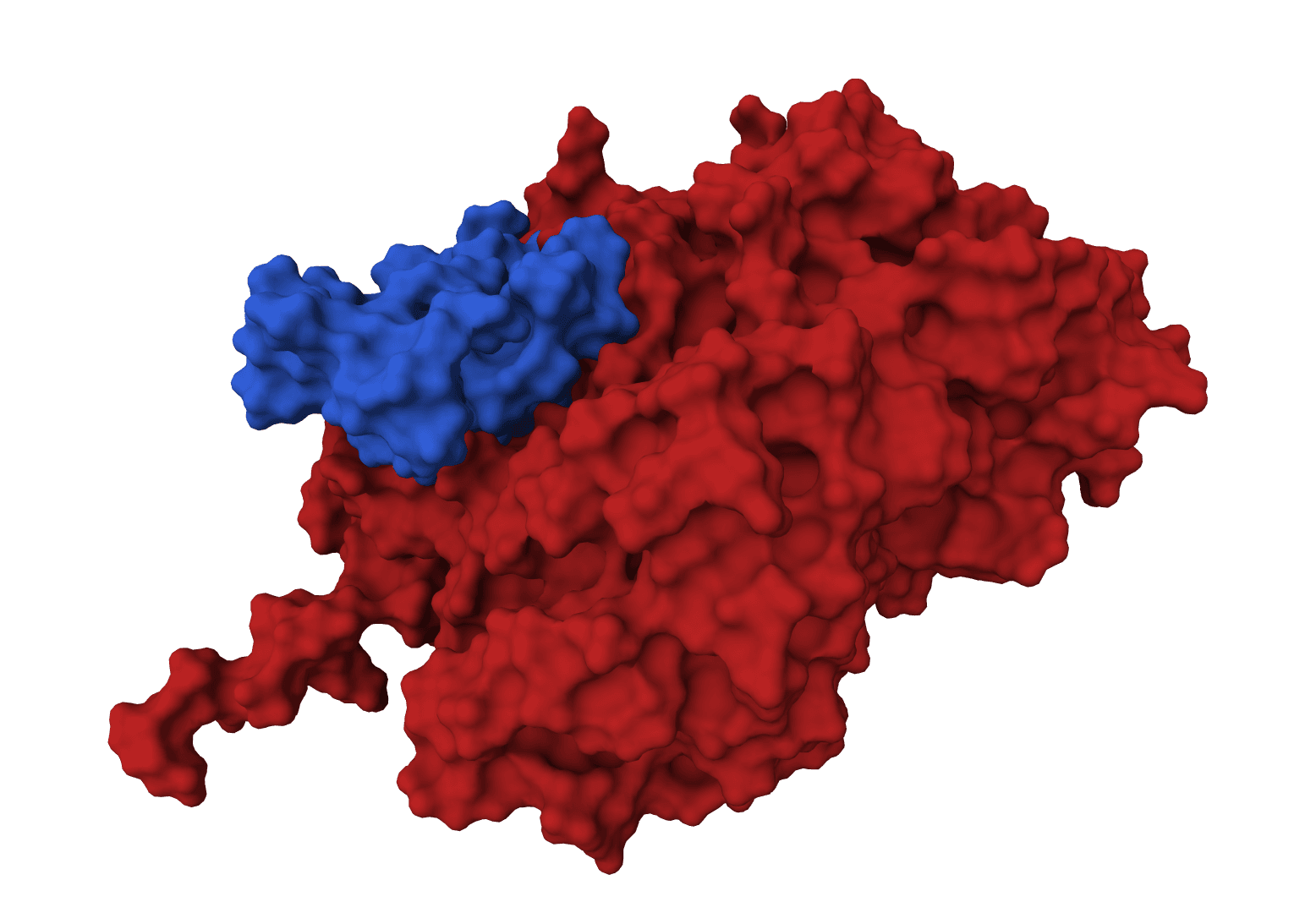
GeoDock predicts flexible protein-protein docking complexes from two separate protein structures using a multi-track iterative transformer and the DIPS 0.3 checkpoint from the Gray Lab release.
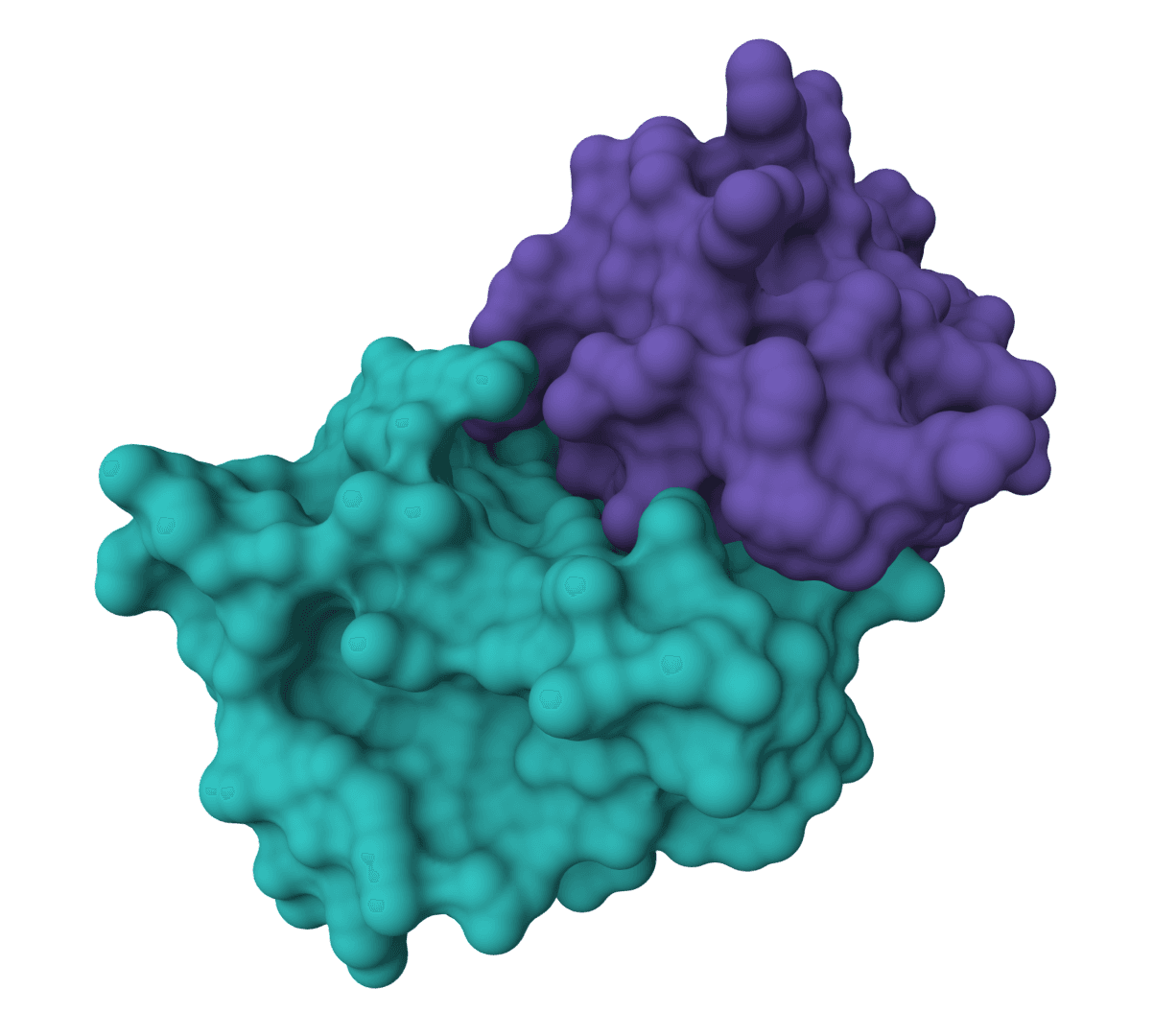
ColabDock is a protein-protein docking framework that uses AlphaFold2 to predict complex structures guided by experimental restraints from cross-linking mass spectrometry, NMR, or other sources.
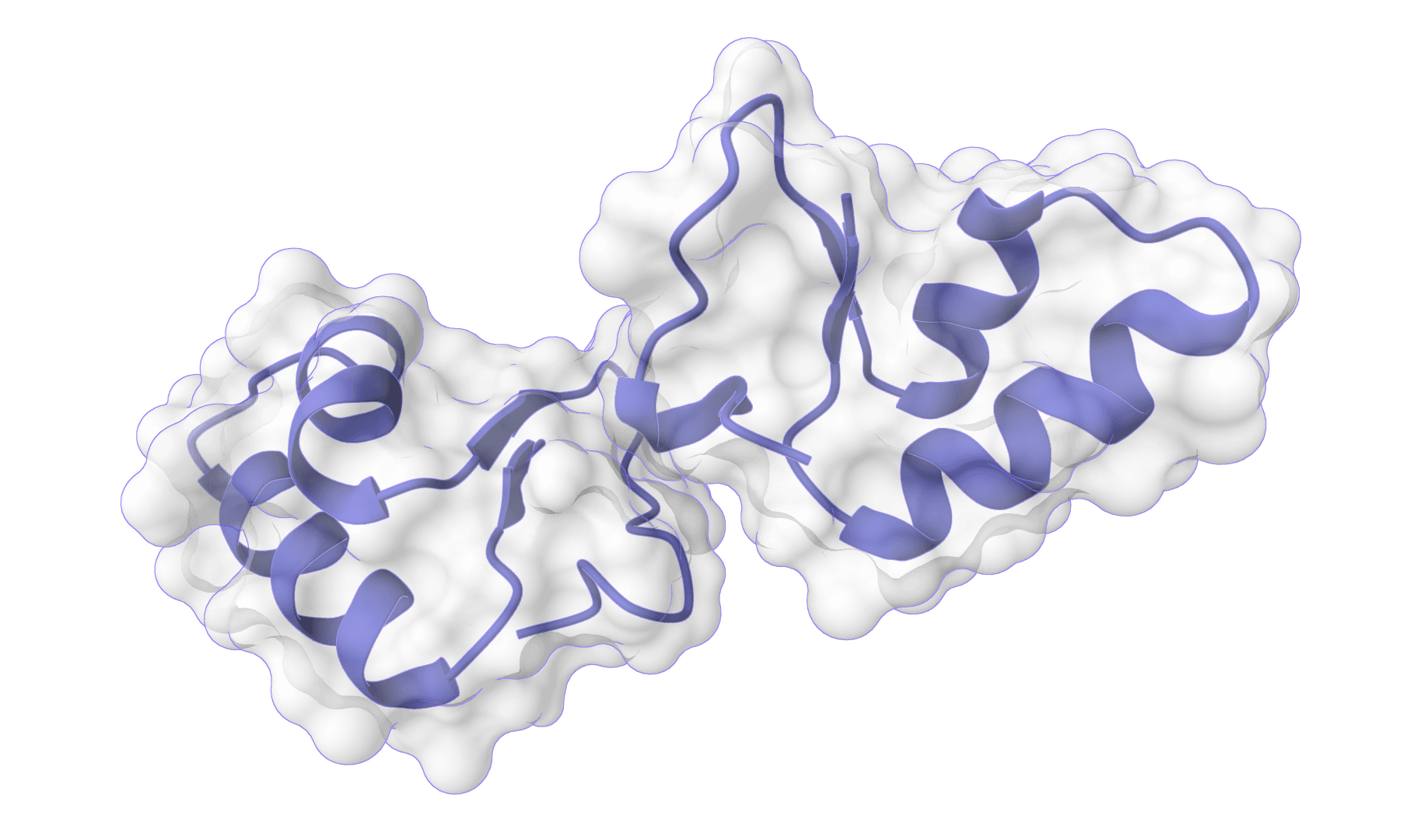
HADDOCK (High Ambiguity Driven protein-protein DOCKing) is an integrative modeling platform for biomolecular complexes. It uses experimental data and bioinformatic predictions to guide the docking process, generating accurate protein-protein complex structures.
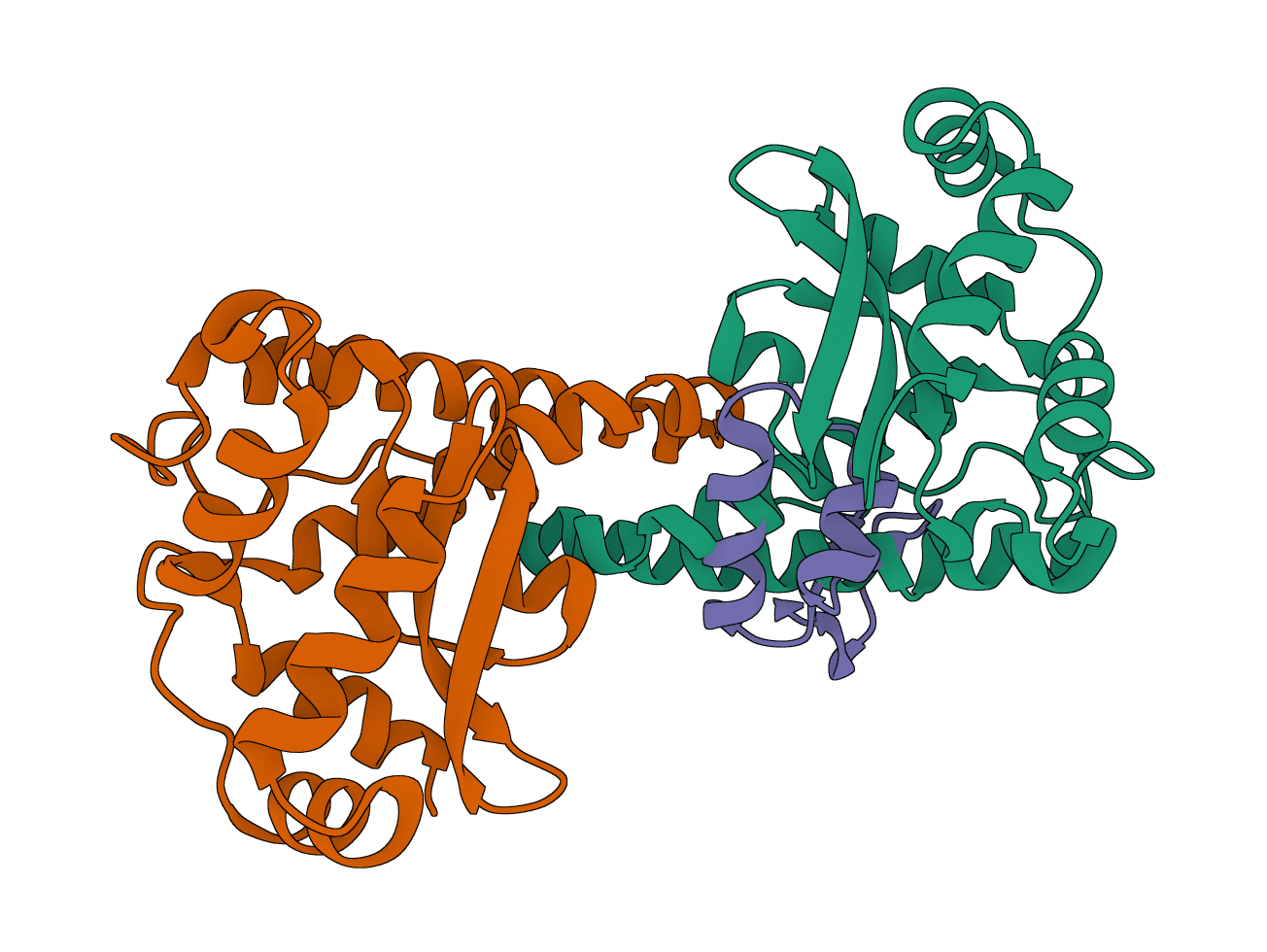
LightDock is a protein-protein, protein-peptide, and protein-DNA docking framework using Glowworm Swarm Optimization (GSO). It predicts macromolecular binding modes and interfaces for biological complexes.
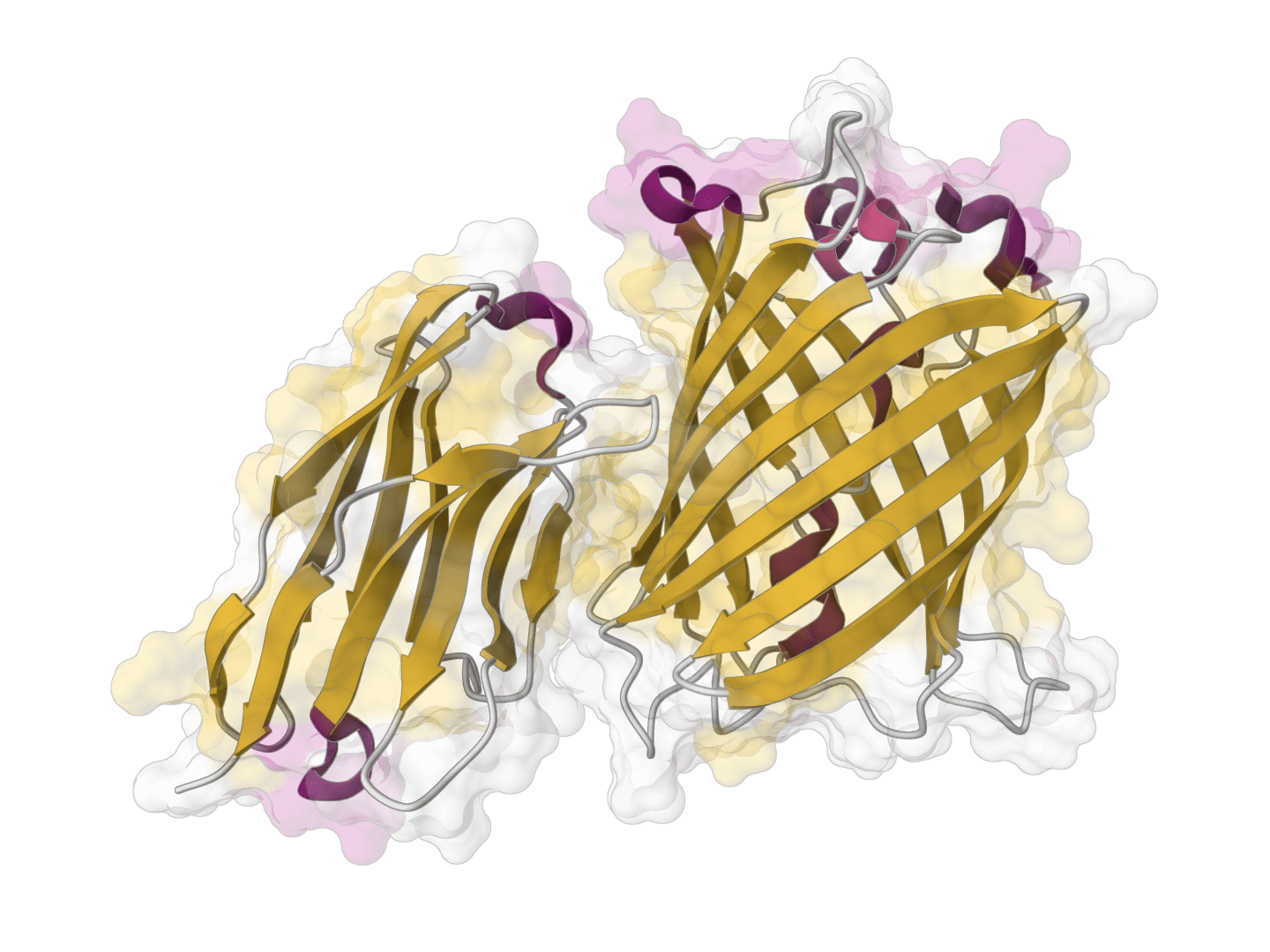
ParaSurf is a state-of-the-art surface-based deep learning model for predicting interactions between antibodies and antigens. It identifies paratope binding sites on antibody structures with high accuracy across multiple benchmark datasets.
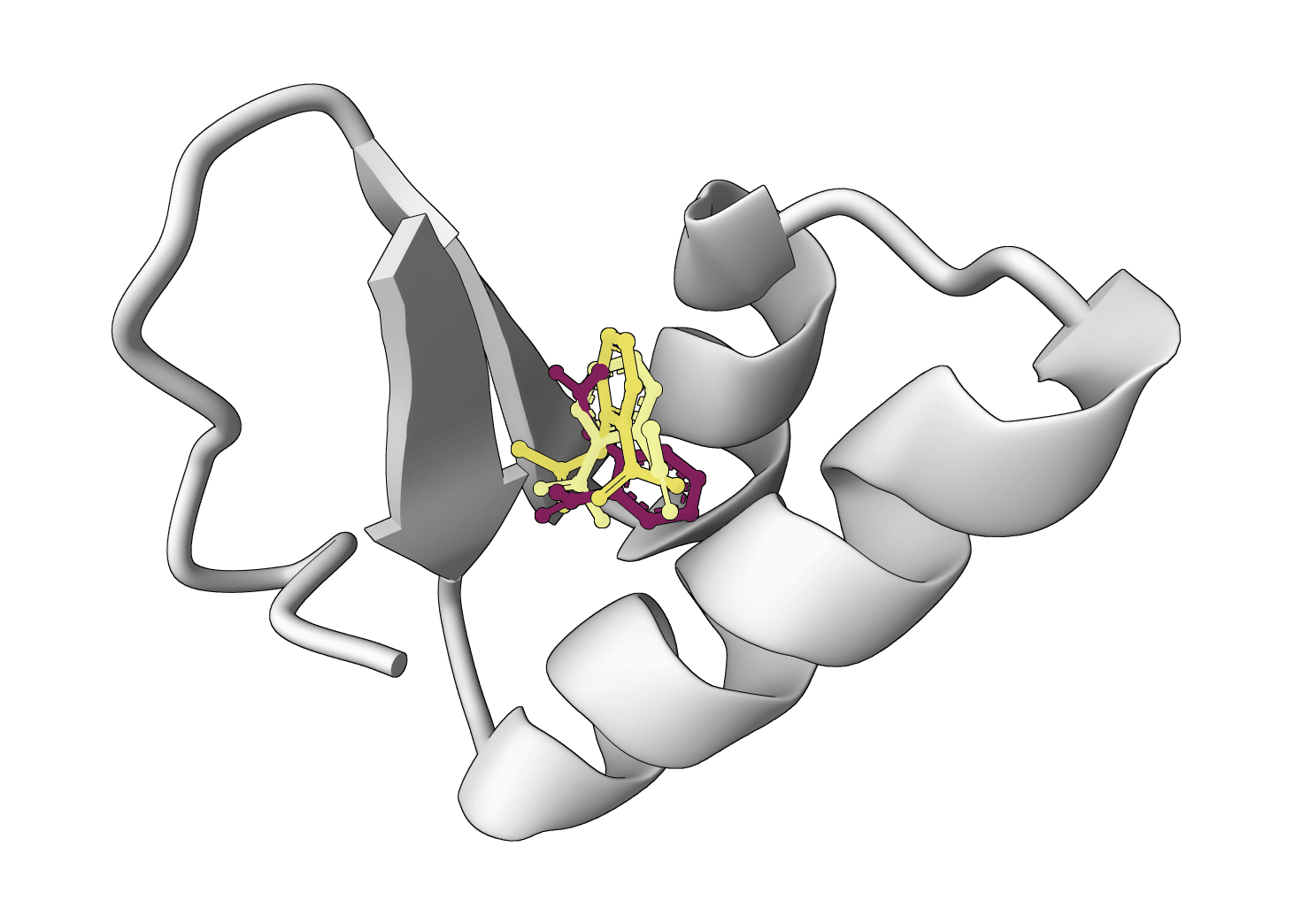
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
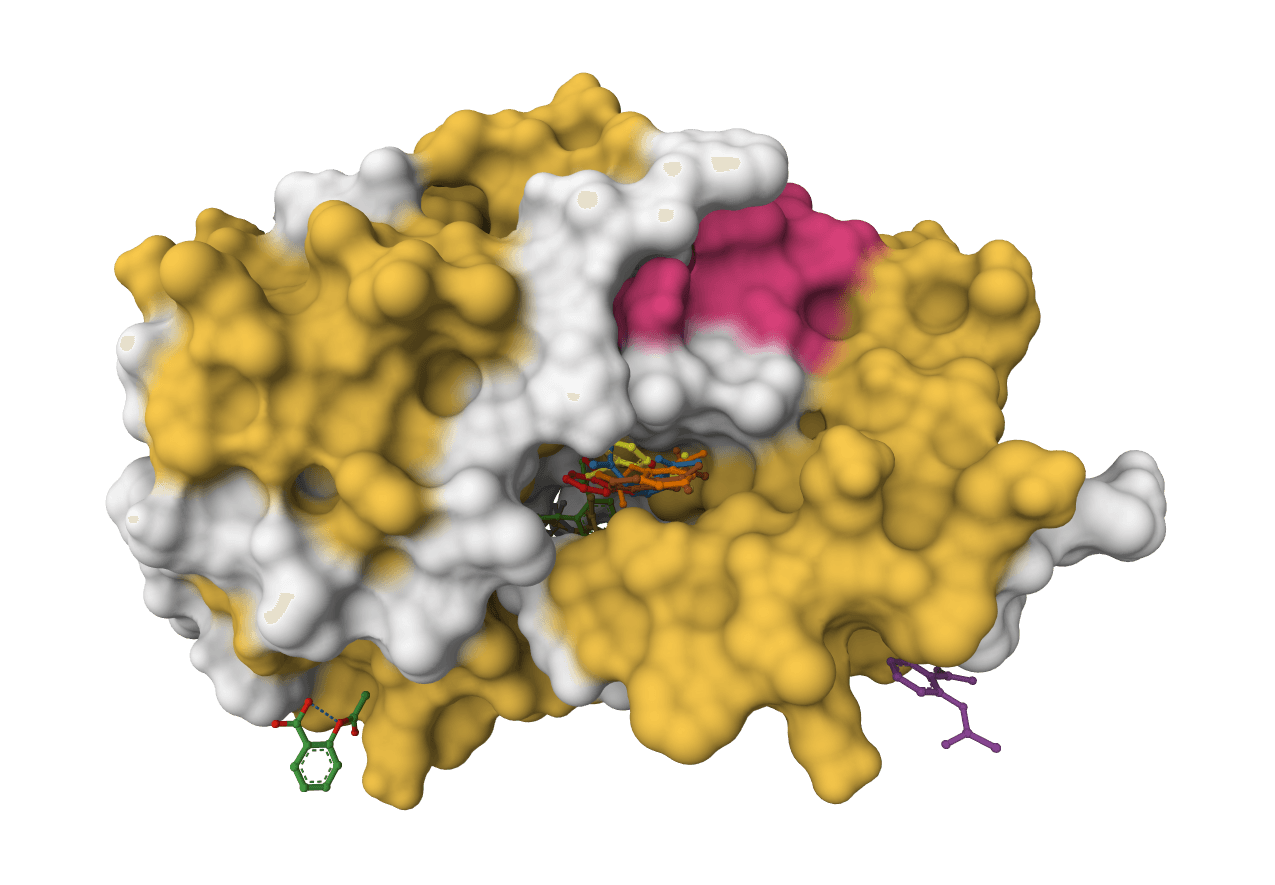
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
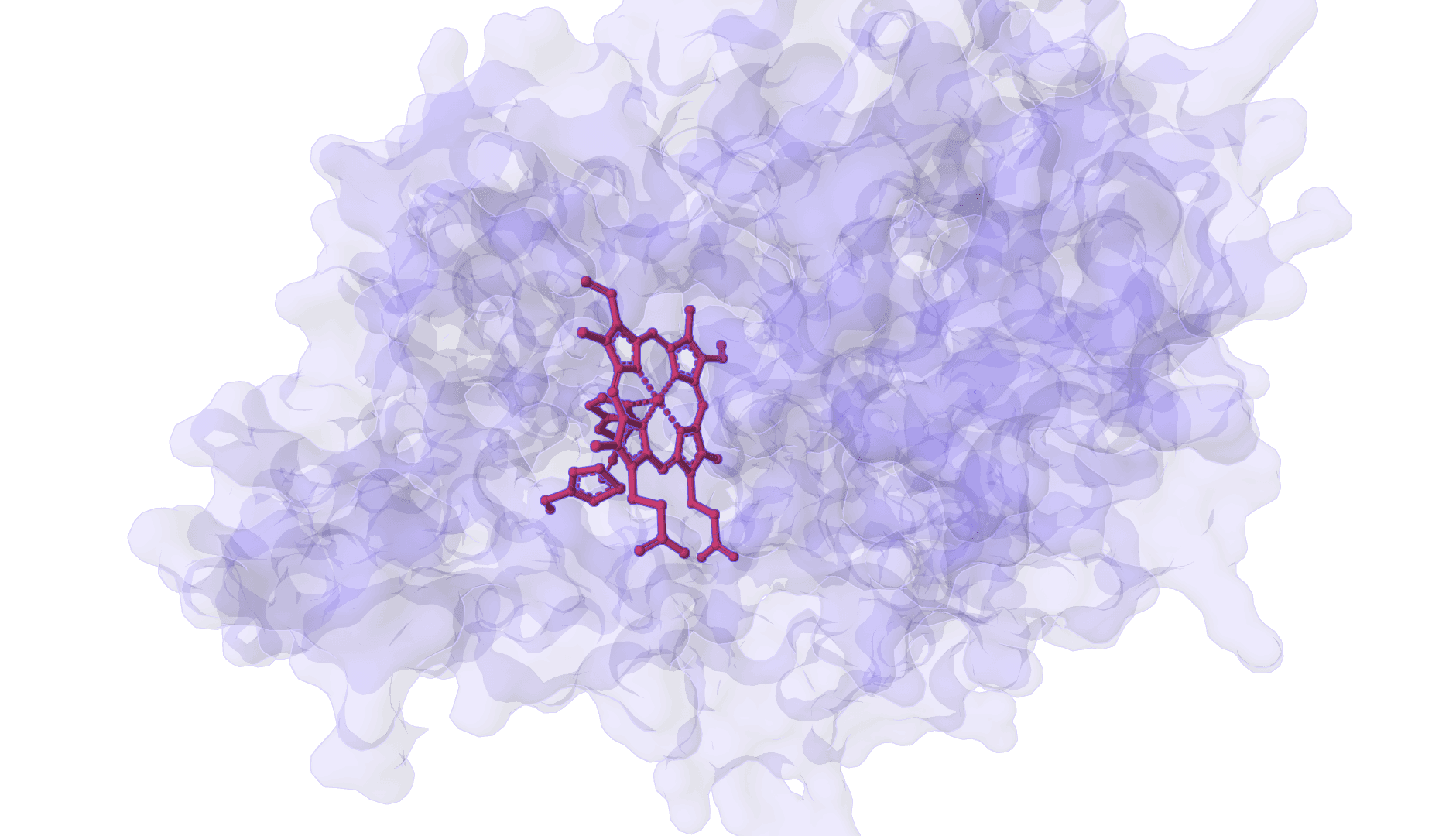
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
AF2Dock is a Gray Lab method for structure-based protein-protein docking. It adapts AlphaFold2-style co-folding to start from two protein structures, then refines receptor-ligand placement through flow matching and ranks sampled complexes with iPTM.
Use AF2Dock when you already have receptor and ligand structures and want a set of ranked complex models without manually defining interface restraints. The default hosted model uses the AF2Dock_base checkpoint.
Upload a receptor protein structure and a ligand protein structure, or fetch each partner from RCSB. AF2Dock accepts .pdb and .cif files. RCSB entries are fetched as CIF structures by default because AF2Dock checks file suffixes directly and supports pdb and cif.
If either structure has unresolved residues that must be mapped back to a full chain sequence, add the optional A3M alignment for that partner. The A3M must include two records for every chain: for chain A, name them A_full and A. Those records must have the same aligned length, and the non-gap positions in the chain record must equal the resolved residue count in the uploaded structure.
| Input | Accepted format | Description |
|---|---|---|
Receptor structure | .pdb, .cif, or RCSB ID | First protein docking partner. Protein ATOM records are required. |
Ligand structure | .pdb, .cif, or RCSB ID | Second protein docking partner moved during docking. Protein ATOM records are required. |
Receptor A3M alignment | .a3m | Optional mapping from resolved receptor residues to full receptor sequence when residues are missing. |
Ligand A3M alignment | .a3m | Optional mapping from resolved ligand residues to full ligand sequence when residues are missing. |
| Setting | Default | Description |
|---|---|---|
Number of samples | 1 | Number of sampled complexes to generate. Increase toward the native 40-pose source setting when you need alternative interfaces. |
Flow-matching steps | 10 | Native --num_steps value. More steps increase refinement time. |
Merge first steps | 0 | Native --merge_first_n_steps value. Leave at 0 for the standard run. |
Additional refine steps | 0 | Native --additional_refine_steps value. Adds refinement after interpolation. |
Random seed | blank | Blank lets AF2Dock choose a random seed. Enter a non-negative integer for reproducibility. |
Filter low-pLDDT residues | false | Leave off to preserve the default behavior of no input pLDDT cutoff. |
ProteinIQ caps hosted jobs at 40 samples, 50 flow-matching steps, 2,200 total residues, and 62 combined receptor-ligand chains so jobs fit the shared GPU runtime and AF2Dock's PDB output format.
AF2Dock writes a target folder for each submitted receptor-ligand pair. ProteinIQ returns every generated file from that folder.
| File pattern | Description |
|---|---|
*_iptm.csv | Ranked sample table sorted by iPTM from high to low. |
*_sN.pdb | Final predicted complex for sample N, with pLDDT values written into the B-factor column. |
*_sN_ori_chain.pdb | Predicted complex with original residue and chain mapping restored when AF2Dock can recover it. |
*_sN_out.pkl | Confidence and auxiliary output dictionary for sample N, including pLDDT, pTM, iPTM, weighted pTM, PAE, final atom positions, and final atom mask. |
*_sN_t0_template.pdb | Initial template written by AF2Dock at the first step. |
The Metrics tab is sorted by iPTM. Higher iPTM generally indicates a stronger model confidence signal for the sampled interface within the same AF2Dock job. Treat iPTM as a ranking signal for poses from one run, not as an experimental affinity measurement.
Start with the top-ranked complex in the 3D viewer, then compare nearby ranks for alternative interfaces. The *_ori_chain.pdb files are useful when you need to trace residues back to original chain IDs, while the *_out.pkl files preserve detailed confidence arrays for offline analysis.
The first-step *_t0_template.pdb file is returned because AF2Dock writes it during inference even when full intermediate output saving is disabled. If you enable intermediate prediction or confidence settings, additional step-level PDB or pickle files may be included.
| Tool | Best fit | Why choose it |
|---|---|---|
AF2Dock | Protein-protein docking from two structures with AF2-style refinement | Returns ranked complex structures and AF2-style confidence outputs. |
| DFMDock | Fast rigid docking without MSAs or AlphaFold-style templates | Useful for exploratory structure-only docking with learned energy ranking. |
| HADDOCK3 | Experimental restraints or interface residues are available | Integrates explicit docking restraints and refinement stages. |
| ColabDock | AlphaFold2-guided docking with manual or reference-derived restraints | Better fit when restraint information should steer complex prediction. |
| DockQ | Evaluate a predicted complex against a known reference | Use after docking when a native or benchmark complex is available. |
| PPAP | Estimate protein-protein affinity for a predicted complex | Use after docking when ranking or triaging protein interfaces by affinity is needed. |
AF2Dock is most useful when receptor and ligand structures are available and the docking question benefits from AlphaFold2-style confidence outputs. It is less suitable for non-protein ligands, nucleic-acid partners, cofactors, large induced-fit motions, or structures whose missing residues cannot be mapped with an A3M file.
Based on Graylab/AF2Dock v1.0.0 and the AF2Dock_base model checkpoint.
DFMDock (Denoising Force Matching Dock) is a diffusion model that unifies sampling and ranking for protein-protein docking within a single framework. It predicts docked poses for protein-protein complexes from unbound structures using denoising score matching with optional clash force guidance.
EquiDock is an SE(3)-equivariant graph neural network for rigid protein-protein docking. It predicts a binding pose for a protein-protein complex from unbound structures using geometric deep learning, with DIPS and DB5 pretrained checkpoints from the native release.
GeoDock predicts flexible protein-protein docking complexes from two separate protein structures using a multi-track iterative transformer and the DIPS 0.3 checkpoint from the Gray Lab release.
ColabDock is a protein-protein docking framework that uses AlphaFold2 to predict complex structures guided by experimental restraints from cross-linking mass spectrometry, NMR, or other sources.
HADDOCK (High Ambiguity Driven protein-protein DOCKing) is an integrative modeling platform for biomolecular complexes. It uses experimental data and bioinformatic predictions to guide the docking process, generating accurate protein-protein complex structures.
LightDock is a protein-protein, protein-peptide, and protein-DNA docking framework using Glowworm Swarm Optimization (GSO). It predicts macromolecular binding modes and interfaces for biological complexes.
ParaSurf is a state-of-the-art surface-based deep learning model for predicting interactions between antibodies and antigens. It identifies paratope binding sites on antibody structures with high accuracy across multiple benchmark datasets.
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.