FASTA splitter
Split large FASTA files by sequence count or export individual files for each sequence.
Input
Output
Configure input settings on the left, then click "Generate"
Split large FASTA files by sequence count or export individual files for each sequence.
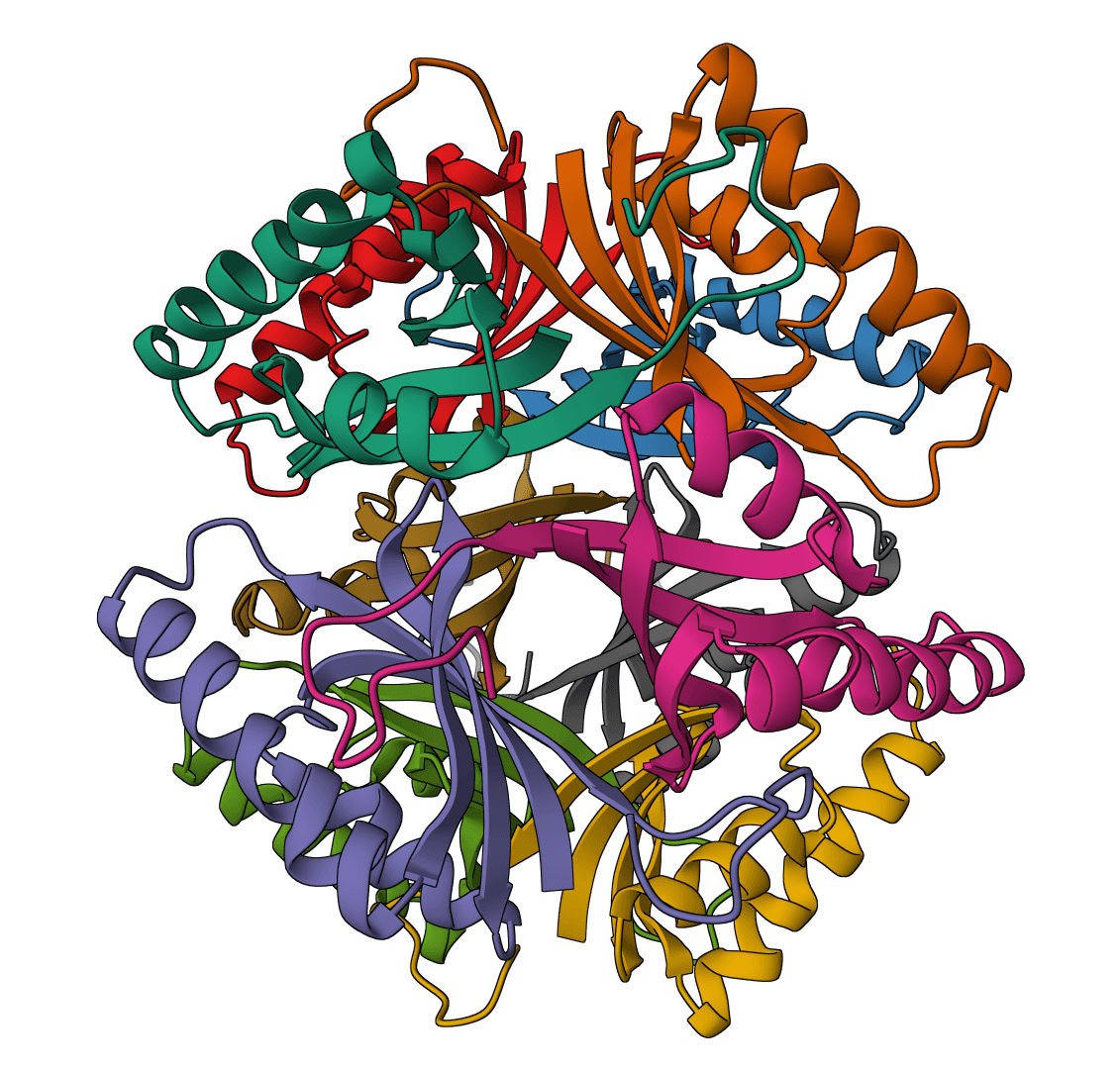
PDB2PQR prepares protein structures for electrostatics calculations by adding missing atoms, predicting protonation states using PROPKA, and assigning atomic charges and radii from standard force fields.
Clean and filter protein sequences by removing or replacing non-standard amino acid characters. Supports multiple filter modes including standard 20 amino acids, IUPAC codes, and custom character sets.
Clean and filter DNA sequences by removing or replacing non-standard nucleotide characters. Supports multiple filter modes including standard 4 bases, IUPAC ambiguity codes, and custom character sets.
Fix ligand files that fail RDKit, Meeko, or docking preparation. Repair SDF, MOL, and MOL2 inputs, apply safe chemistry cleanup, and export docking-ready SDF files.
Convert CSV and TSV files containing sequence data to FASTA format with flexible column mapping and automatic delimiter detection

Convert TXT or plain text sequences into FASTA format files for DNA, RNA, and protein workflows with cleanup, validation, and downloads
Extract sequence features (CDS, mRNA, gene, etc.) from GenBank files in FASTA format with support for spliced features
Convert FASTA sequence files to FASTQ format with mock quality scores
Convert FASTQ sequence files to FASTA format
Convert GenBank files to FASTA format
FASTA Splitter divides multi-sequence FASTA files into smaller, manageable pieces. This is useful when downstream tools have sequence limits, when you need to parallelize analysis across multiple files, or when organizing sequences for batch processing.
Many bioinformatics tools restrict the number of sequences per submission. Instead of manually copying sequences into separate files, this tool automates the splitting process while preserving headers and sequence integrity.
The tool parses your input FASTA file, identifies individual sequences by their header lines (starting with >), and distributes them into output files according to your chosen split mode.
All processing happens in your browser—your sequences are never uploaded to a server. The tool generates a ZIP archive containing all split files, ready for download.
Split by sequence count groups a fixed number of sequences per file. Individual files creates one file per sequence. Split by line treats each line as a separate file (rarely needed).Split by sequence count mode, this controls how many sequences go into each output file.Output files need consistent, predictable names for downstream automation.
Numbered (part_001, part_002), Sequential (file_1, file_2), or Header-based (uses the first sequence's header as the filename).The tool produces a ZIP archive containing:
File extensions match your input (.fasta, .fa, or .fas).
| Scenario | Recommended settings |
|---|---|
| Preparing for AlphaFold (1 sequence per job) | Split mode: Individual files |
| Batch processing with 100-sequence limit | Split mode: Split by sequence count, Sequences per file: 100 |
| Organizing sequences by identity | Split mode: Individual files, Naming: Header-based |
We recommend keeping Preserve original headers enabled unless you have a specific reason to modify them. Header information often contains identifiers needed to trace results back to source sequences.
For large files with hundreds of sequences, Numbered naming provides cleaner organization than Header-based, which can produce long or problematic filenames from complex headers.