
15 credits
Configure input settings on the left, then click "Submit"orLoad an example (it's free)
(Single mode) HIV-1 Protease—Saquinavir
(Simultaneous co-docking) BRD4 Bromodomain—Fragments
(Batch mode) SARS-CoV-2 Mpro—Antiviral Repurposing Panel
(Single mode) HIV-1 Protease—Saquinavir
(Simultaneous co-docking) BRD4 Bromodomain—Fragments
(Batch mode) SARS-CoV-2 Mpro—Antiviral Repurposing Panel
GPU-accelerated molecular docking using the AutoDock4 force field. Up to 56x faster than serial AutoDock via CUDA parallelization of the Lamarckian Genetic Algorithm.
Open-source molecular docking platform using physics-based scoring functions. CPU-optimized algorithms achieve sub-angstrom accuracy (0.014A RMSD) without GPU requirements.
SMINA is a fork of AutoDock Vina with enhanced scoring functions, custom scoring support, and 10-20x faster minimization. Ideal for scoring function development, pose refinement, and high-performance docking workflows.
GNINA is a molecular docking tool that combines traditional physics-based docking with deep learning CNN scoring for protein-small-molecule complexes. It provides accurate binding predictions with confidence scores, optimized for high-throughput virtual screening.
DiffDock-L is a state-of-the-art molecular docking tool that uses diffusion models to predict how small molecule ligands bind to protein targets. It generates multiple binding poses with confidence scores.
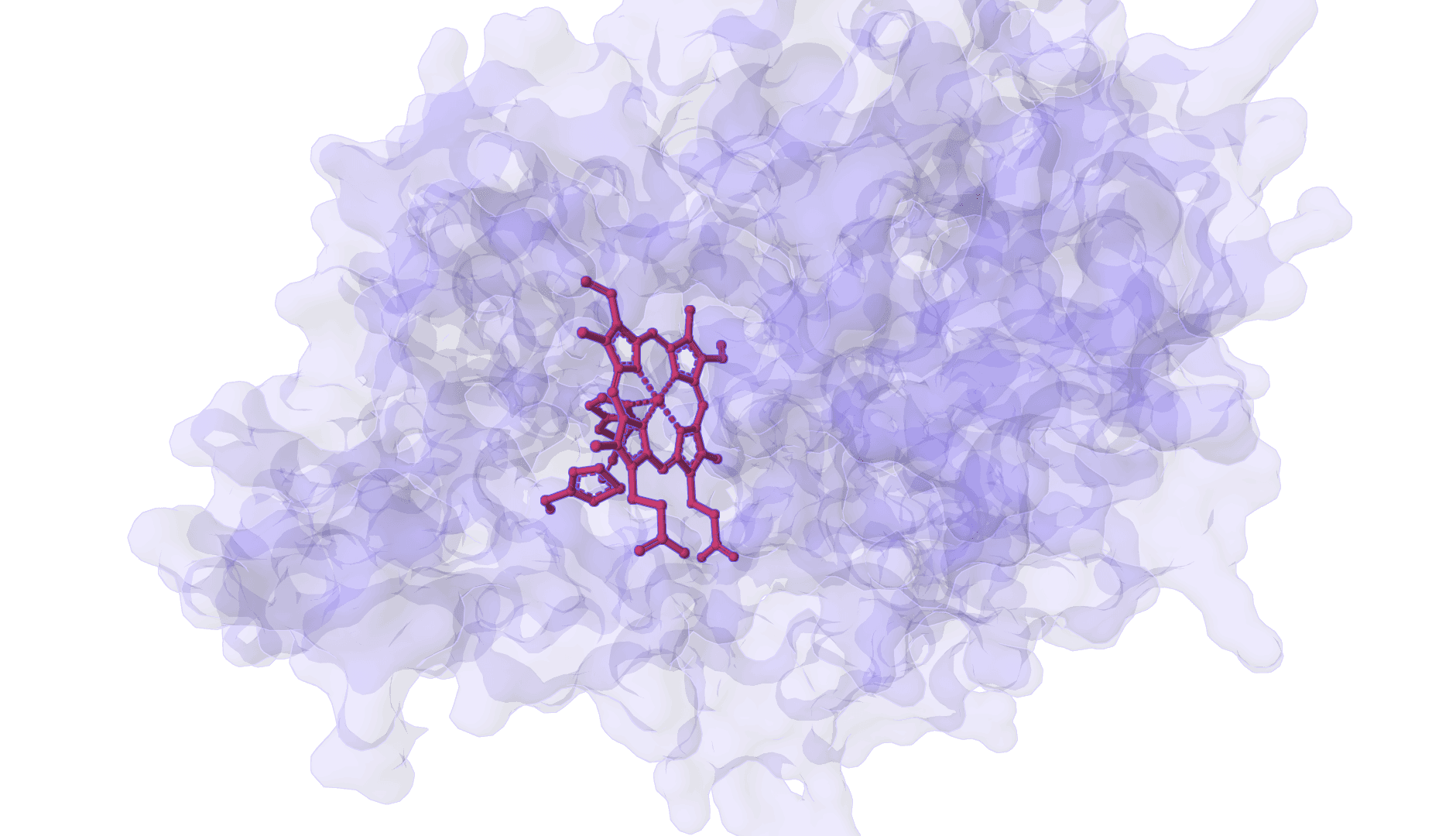
DynamicBind is an AI-powered protein-ligand binding prediction tool that recovers ligand-induced conformational changes from unbound protein structures. It predicts both ligand binding poses and protein conformational changes.
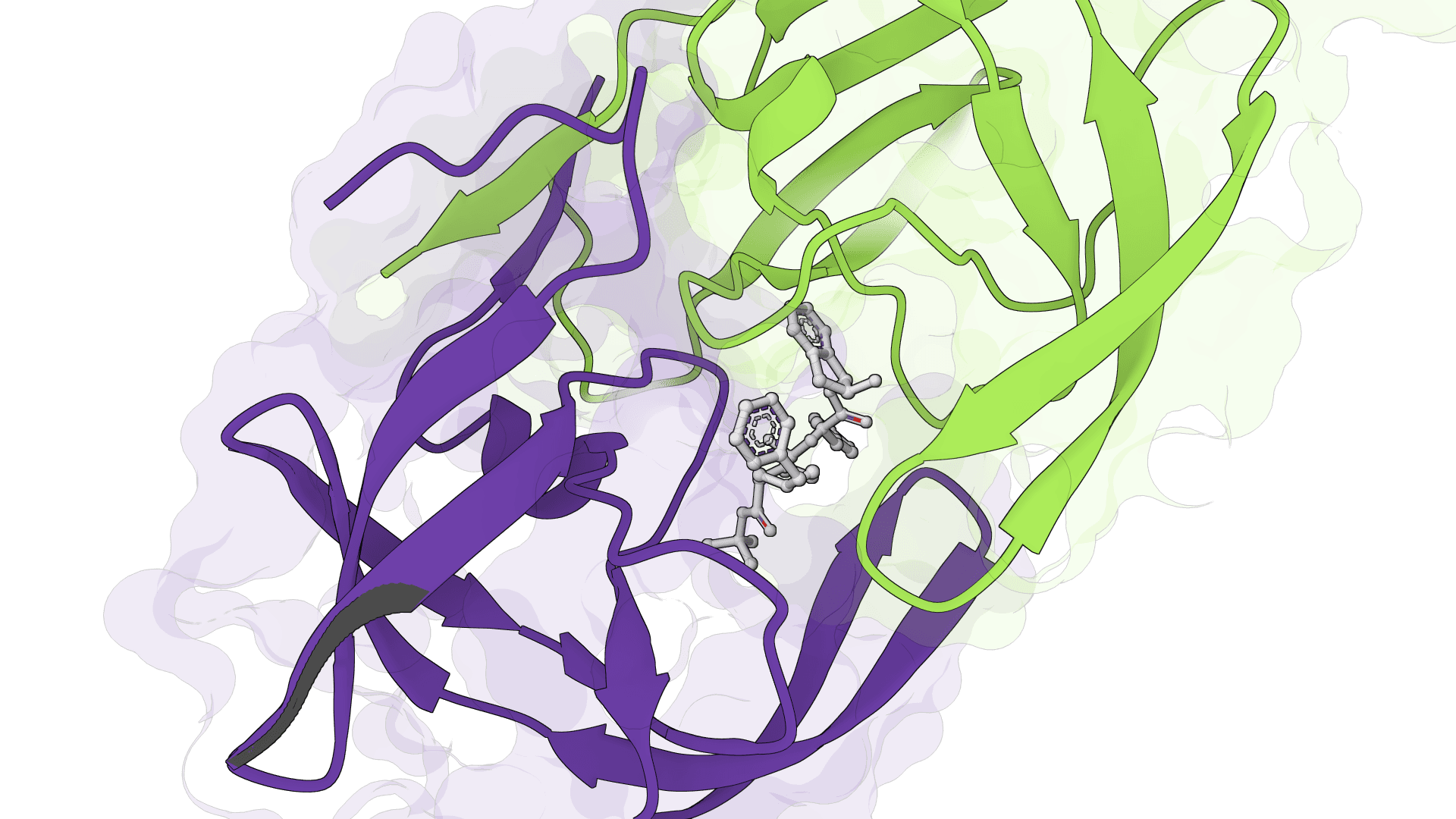
SigmaDock is a fragment-based molecular docking tool using SE(3) equivariant diffusion models to predict how small molecule ligands bind to protein targets. Presented at ICLR 2026, it generates multiple binding poses with Vinardo scoring.
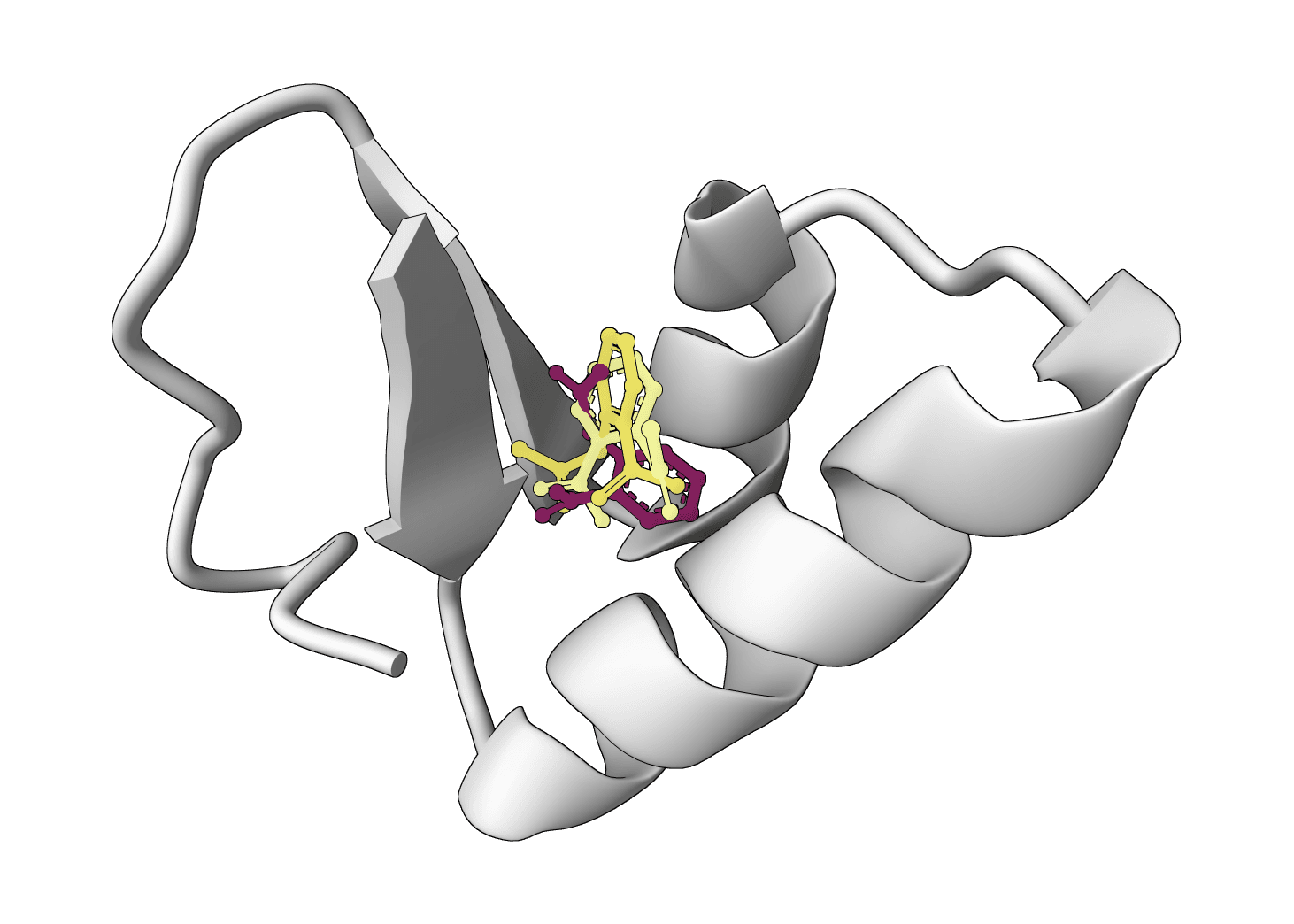
SurfDock is a surface-informed diffusion generative model for protein-ligand docking, published in Nature Methods 2024. It leverages protein surface geometry to guide a diffusion process for reliable and accurate protein-ligand complex prediction.
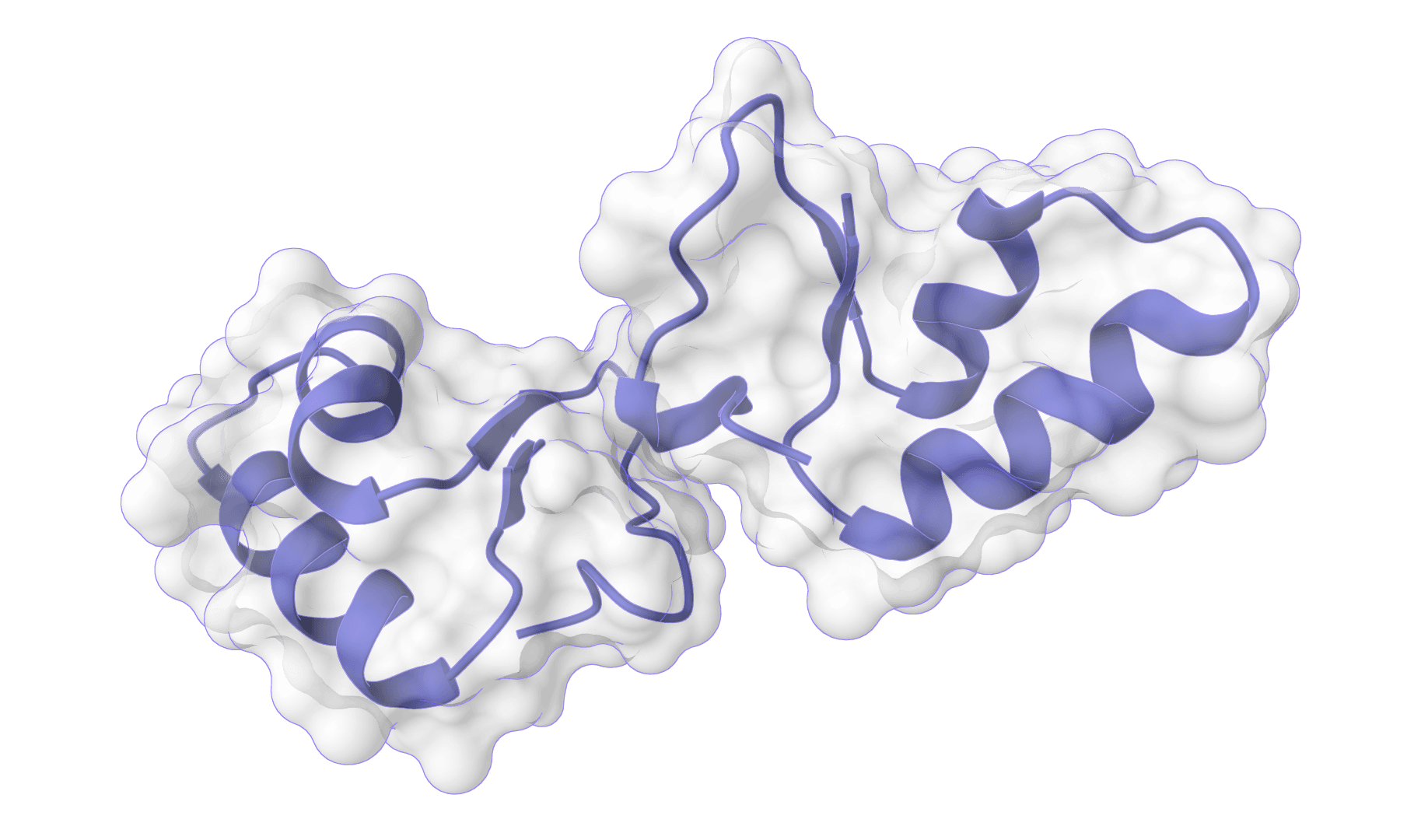
HADDOCK (High Ambiguity Driven protein-protein DOCKing) is an integrative modeling platform for biomolecular complexes. It uses experimental data and bioinformatic predictions to guide the docking process, generating accurate protein-protein complex structures.
LightDock is a protein-protein, protein-peptide, and protein-DNA docking framework using Glowworm Swarm Optimization (GSO). It predicts macromolecular binding modes and interfaces for biological complexes.
AutoDock Vina is a molecular docking program that predicts how a small molecule binds to a protein by searching through possible binding poses and scoring them with an empirical force field. It is the most widely used open-source docking tool, combining fast search with practical accuracy for routine structure-based drug discovery.
On ProteinIQ, the Vina interface supports the full Vina family workflow rather than just the original 2010 defaults. This includes the standard Vina scoring function, the Vinardo variant optimized for virtual screening, and an AutoDock4 scoring option for metal coordination or legacy workflows.
ProteinIQ runs AutoDock Vina in the browser with cloud execution, so receptor preparation, ligand submission, and docking outputs are available without local installation or command-line setup.
| Input | Description |
|---|---|
Protein (Receptor) | Upload a receptor structure as .pdb or .ent, or fetch one by 4-character PDB ID such as 1HSG. Protein atoms are required, and the validator warns when the uploaded file appears to contain nucleic acids or YASARA-specific formatting. |
Ligand | Provide ligand input as SMILES text, a supported structure file (.pdbqt, .sdf, .mol, .mol2, .smiles, .smi, .txt, .csv), or fetch by PubChem. One ligand is used in Single ligand mode, while Simultaneous co-docking accepts up to 5 ligands in one run. |
Vina docking mode | Single ligand runs one docking job, while Simultaneous co-docking places multiple ligands in the same search space during one run. |
Job name | Optional label stored with the run to make repeated docking experiments easier to identify. |
Ligand validation blocks metal-containing ligands and disconnected multi-fragment submissions in the standard Vina workflow. Those cases are better suited to GNINA or to a more specialized docking setup.
| Setting | Description |
|---|---|
Scoring function | Vina is the default general-purpose choice. Vinardo uses a modified empirical model often favored for virtual screening benchmarks. AutoDock4 uses the classical AutoDock4 force-field-style scoring and is the required option for hydrated ligand and zinc-specific workflows in this interface. |
Docking mode | Dock performs a full search. Score only evaluates the submitted pose without searching. Local only refines the input pose locally. Randomize only generates randomized ligand placement without full docking output ranking. |
Exhaustiveness | Search thoroughness from 1 to 64, with a default of 8. Larger values increase runtime but improve the chance of recovering lower-energy poses. |
Number of poses | Maximum number of docked poses returned, from 1 to 50. |
Energy range (kcal/mol) | Retains poses within the specified energy window from the best-ranked pose. Larger values preserve more alternative solutions. |
Min RMSD between poses (Å) | Minimum structural separation between reported poses. Increasing this value reduces near-duplicate outputs. |
Max evaluations | Caps scoring function evaluations. 0 leaves the choice to Vina's automatic internal heuristic. |
Random seed | Integer seed for reproducible searches. 0 allows nondeterministic initialization. |
Search mode | Auto builds a search region around the receptor, Manual supports explicit center and size fields, and Autobox generates a receptor-centered box with configurable padding. |
Center X, Y, Z | Coordinates of the search-box center in Manual mode. |
Size X, Y, Z | Search-box dimensions in Å. Smaller boxes are faster and usually more reliable when the binding site is already known. |
Auto-box padding (Å) | Extra padding added around the automatically determined search region. |
Grid spacing (Å) | Spacing used for affinity map discretization. Smaller spacing increases resolution at higher computational cost. |
Force even voxels | Rounds the box definition to an even grid count for more controlled map geometry. |
Flexible residues |
| Output | Description |
|---|---|
Viewer | Interactive 3D view of receptor and docked pose geometry |
Data | Table of scored poses with binding affinity (kcal/mol) and links to output files |
Files | Downloadable structure files for the generated docking poses |
AutoDock Vina combines an empirical scoring function with stochastic global search and gradient-based local optimization. The ligand is translated, rotated, and flexed inside a predefined search volume; each candidate pose is scored; and promising conformations are refined before final clustering and ranking.
The scoring function estimates binding favorability using weighted steric, hydrophobic, hydrogen-bonding, and penalty terms. Vina uses the default empirical model. Vinardo modifies the parameterization for improved enrichment in some screening benchmarks. AutoDock4 uses older force-field-style terms and is required when metal-aware behavior or hydrated ligand workflows are needed.
Vina uses multiple independent runs initialized with random poses. Within each run it alternates broad perturbation steps with local refinement, then collects the best solutions across all searches. The Exhaustiveness parameter controls total independent search effort, while Number of poses and Energy range determine how many alternatives survive to the final report.
Most docking calculations keep the receptor rigid apart from ligand torsions. When Flexible residues are specified, selected receptor side chains are allowed to move during the search. This can recover poses that rigid docking would miss, but increases the search space and runtime substantially. Flexible docking is usually reserved for a few residues with a clear mechanistic rationale for moving.
The affinity score is most useful as a relative ranking within one consistent experiment. Absolute kcal/mol values should not be treated as direct binding free energies, especially across different targets, protonation states, or receptor preparations.
| Affinity range | Typical interpretation |
|---|---|
| < -10 kcal/mol | Very favorable predicted binding |
| -7 to -10 kcal/mol | Strong docking score worth follow-up |
| -5 to -7 kcal/mol | Moderate score that often needs visual inspection |
| > -5 kcal/mol | Weak or uncertain binding hypothesis |
Pose geometry matters as much as score. A slightly worse score with sensible hydrogen bonding, steric fit, and ligand burial is often more credible than the top-ranked pose if that pose shows clashes or unrealistic exposure. For batch docking, comparisons are most meaningful when all ligands were prepared with the same protonation and tautomer assumptions.
Comma-separated residues such as A:315,B:42 to model selected side-chain flexibility during docking. |
Hydrated ligand workflow | Treats explicit ligand-associated waters as part of the docking setup. Available only with AutoDock4 scoring. |
Zinc metalloprotein mode | Uses zinc-focused receptor preparation assumptions. Available only with AutoDock4 scoring. |
Disable post-docking refinement | Skips the final local refinement step for faster turnaround at some cost to pose quality. |
Use custom scoring weights (advanced) | Unlocks manual overrides for the weight coefficients used by Vina, Vinardo, or AutoDock4. |