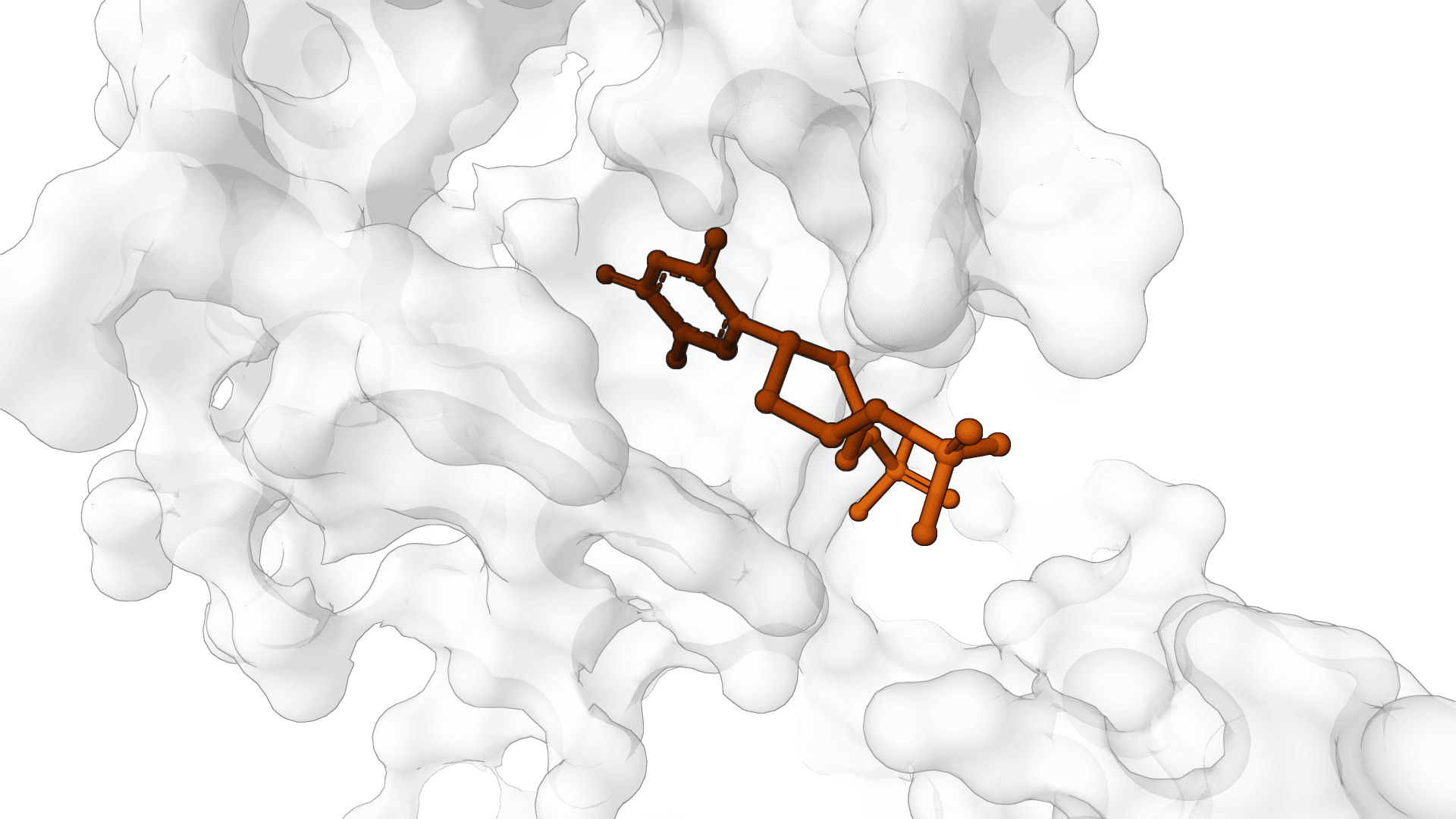
AF2BIND finds the residues on a protein most likely to contact a small-molecule ligand. It reads a single protein structure and returns a per-residue p(bind) score, so the binding pocket shows up as a cluster of high-scoring residues even when no ligand is present in the input.
The method works by borrowing AlphaFold2's learned sense of which residues sit at interfaces. Instead of docking an actual molecule, it presents the protein with a 20-residue bait made from single amino acids acting as stand-in ligands, then reads how strongly AlphaFold2's internal representation couples each target residue to those probes. Residues that the network treats as interface-like score high. This makes AF2BIND a fast way to localize a pocket before committing to docking or pocket geometry analysis.
AF2BIND scores one chain or compact domain at a time. ProteinIQ accepts proteins up to 300 residues; for anything larger, trim to the domain or interface region of interest before scoring.
Upload a protein structure as a PDB file or fetch one from RCSB by ID, pick the chain to score, and AF2BIND returns a ranked table of p(bind) values plus a copy of the structure colored by those scores. The prediction runs on GPU infrastructure with no AlphaFold2 setup, no MSA database, and no local install. The top-ranked residues mark the most probable ligand-binding site.
| Input | Description |
|---|---|
Target protein | A single PDB file or an RCSB PDB ID. Must contain protein atoms, up to 300 residues. |
| Setting | Description |
|---|---|
Target chain | Chain to score, by default A. Only one chain is scored per run. |
Mask sidechains | Hides target sidechain atoms from the model so scoring leans on backbone geometry. On by default, matching the AF2BIND default rm_target_sc=True. |
Mask sequence | Hides the target sequence identity so scoring leans on structure alone. Off by default. |
Rescale by maximum p(bind) | Normalizes the p(bind) values written into the structure so the highest-scoring residue maps to the top of the color range. Off by default, which preserves raw scores for coloring. |
Masking sidechains and keeping the sequence visible is the configuration AF2BIND was validated with. Turning on sequence masking is useful when testing how much a prediction depends on structure versus sequence, but it is not the recommended starting point.
| Output | Contents |
|---|---|
results.csv | One row per residue with rank, chain, residue number, amino acid, and p(bind). Sorted from highest to lowest score. |
output.pdb | The input structure with p(bind) written into the B-factor column, ready to color in a structure viewer. |
output.zip | The full prediction bundle for download. |
AlphaFold2 builds a pair representation, a tensor that encodes the relationship between every pair of residues across all chains in a complex. When two chains form an interface, the pair representation between their residues carries a distinctive signal that the network learned from real protein structures.
AF2BIND exploits this without needing a real ligand. It adds 20 separate single-residue "bait" chains to the prediction, one for each amino acid type, using the bait sequence ACDEFGHIKLMNPQRSTVWY. Each bait acts as a minimal pseudo-ligand. AlphaFold2 then produces a pair representation linking every target residue to every bait. A small trained logistic-regression layer reads those cross-chain features and outputs p(bind), the probability that a given target residue lies in a ligand-binding pocket.
Because the baits are individual amino acids rather than a specific compound, p(bind) flags where ligands tend to bind in general, not the affinity of any particular molecule.
p(bind) is a ranking signal, not a calibrated probability with a universal cutoff. The pocket reveals itself as a tight group of high-scoring residues that sit close together in 3D, not as isolated high values scattered across the sequence.
A practical reading approach:
p(bind) and inspect the top 10 to 15 residues. In the AF2BIND benchmarks, this top slice captures the binding site for most single-pocket proteins.output.pdb. Residues that cluster spatially are the predicted pocket; lone high scorers far from the cluster are usually noise.For proteins with more than one pocket, expect more than one spatial cluster. The ranking alone will not separate them, so the structural view matters.
AF2BIND is strongest when a structure exists and the goal is to localize a small-molecule pocket quickly, especially for a protein where no bound ligand has been resolved. It needs no MSA and no docking box.
If the structure itself is uncertain, predict it first with AlphaFold 2, then score the model with AF2BIND.
Rank a compound library against one protein target with SPRINT protein and ligand co-embeddings and native cosine similarity.
Deep learning framework for predicting small molecule-RNA interactions using RNA secondary structure. Combines language models, CNNs, and graph attention networks for binding prediction.
Predict ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) properties from SMILES strings using machine learning models trained on Therapeutics Data Commons datasets.
Predict 22 ADMET properties from SMILES strings with the native Admetica Chemprop models from Datagrok.
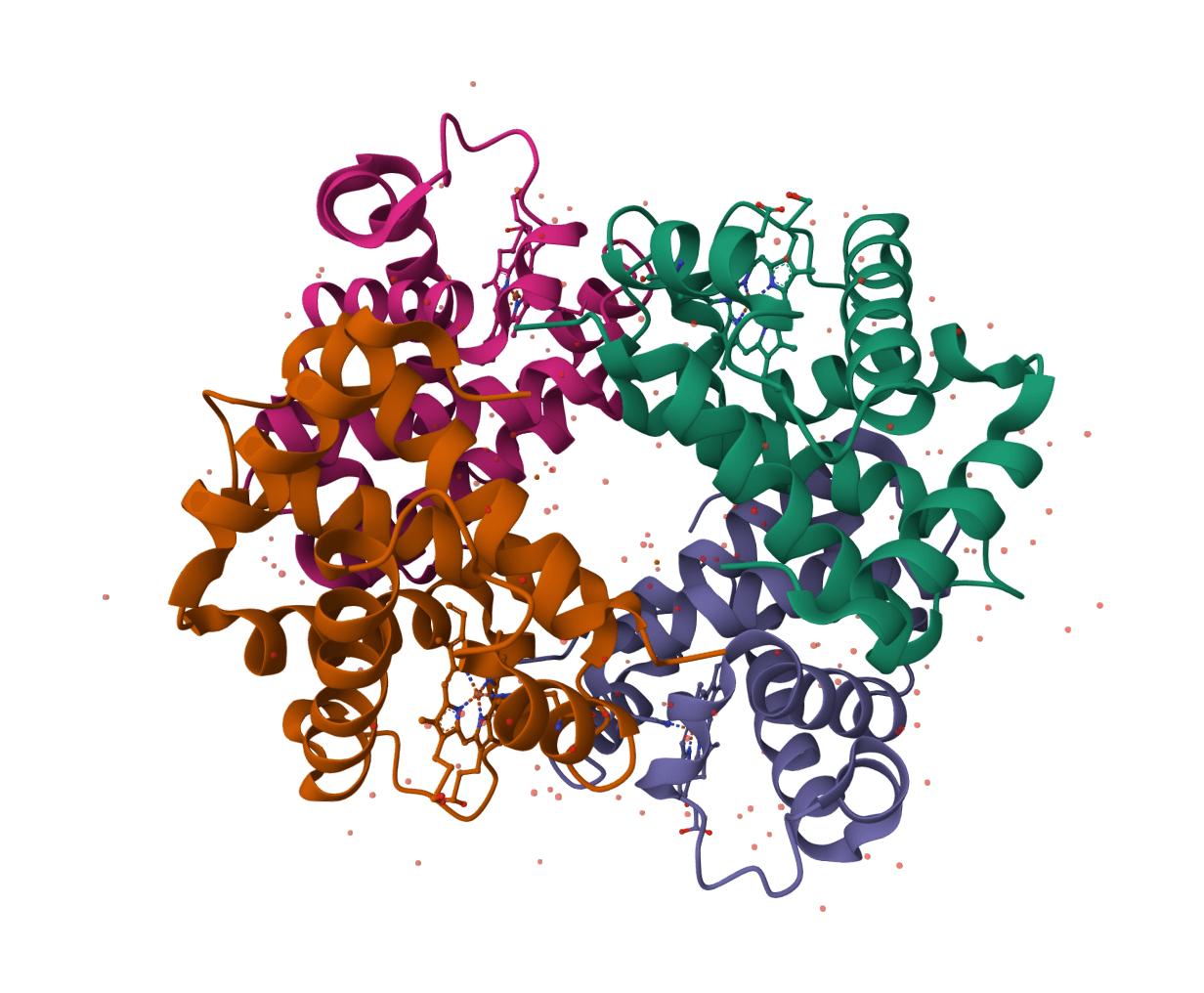
Identify toxic, reactive, and pharmacokinetically problematic molecular fragments using structural alert patterns
Predict toxicity and synthetic accessibility of small molecules using machine learning. eToxPred combines toxicity risk assessment with synthetic accessibility scoring to help prioritize drug candidates.
Screen for lead-like compounds using stricter molecular descriptor criteria than Lipinski or Veber rules for early-stage drug discovery
Screen compounds for Pan-Assay Interference patterns that cause false positives in biological assays
Quantitative estimate for protein-protein interaction inhibitor potential. Evaluates drug-likeness for compounds targeting PPIs.
Screen compounds for structural toxicity alerts using PAINS, Brenk, and NIH filters. For focused screening, see PAINS Filter, Brenk Filter, or Veber's Rule.
AF2BIND finds the residues on a protein most likely to contact a small-molecule ligand. It reads a single protein structure and returns a per-residue p(bind) score, so the binding pocket shows up as a cluster of high-scoring residues even when no ligand is present in the input.
The method works by borrowing AlphaFold2's learned sense of which residues sit at interfaces. Instead of docking an actual molecule, it presents the protein with a 20-residue bait made from single amino acids acting as stand-in ligands, then reads how strongly AlphaFold2's internal representation couples each target residue to those probes. Residues that the network treats as interface-like score high. This makes AF2BIND a fast way to localize a pocket before committing to docking or pocket geometry analysis.
AF2BIND scores one chain or compact domain at a time. ProteinIQ accepts proteins up to 300 residues; for anything larger, trim to the domain or interface region of interest before scoring.
Upload a protein structure as a PDB file or fetch one from RCSB by ID, pick the chain to score, and AF2BIND returns a ranked table of p(bind) values plus a copy of the structure colored by those scores. The prediction runs on GPU infrastructure with no AlphaFold2 setup, no MSA database, and no local install. The top-ranked residues mark the most probable ligand-binding site.
| Input | Description |
|---|---|
Target protein | A single PDB file or an RCSB PDB ID. Must contain protein atoms, up to 300 residues. |
| Setting | Description |
|---|---|
Target chain | Chain to score, by default A. Only one chain is scored per run. |
Mask sidechains | Hides target sidechain atoms from the model so scoring leans on backbone geometry. On by default, matching the AF2BIND default rm_target_sc=True. |
Mask sequence | Hides the target sequence identity so scoring leans on structure alone. Off by default. |
Rescale by maximum p(bind) | Normalizes the p(bind) values written into the structure so the highest-scoring residue maps to the top of the color range. Off by default, which preserves raw scores for coloring. |
Masking sidechains and keeping the sequence visible is the configuration AF2BIND was validated with. Turning on sequence masking is useful when testing how much a prediction depends on structure versus sequence, but it is not the recommended starting point.
| Output | Contents |
|---|---|
results.csv | One row per residue with rank, chain, residue number, amino acid, and p(bind). Sorted from highest to lowest score. |
output.pdb | The input structure with p(bind) written into the B-factor column, ready to color in a structure viewer. |
output.zip | The full prediction bundle for download. |
AlphaFold2 builds a pair representation, a tensor that encodes the relationship between every pair of residues across all chains in a complex. When two chains form an interface, the pair representation between their residues carries a distinctive signal that the network learned from real protein structures.
AF2BIND exploits this without needing a real ligand. It adds 20 separate single-residue "bait" chains to the prediction, one for each amino acid type, using the bait sequence ACDEFGHIKLMNPQRSTVWY. Each bait acts as a minimal pseudo-ligand. AlphaFold2 then produces a pair representation linking every target residue to every bait. A small trained logistic-regression layer reads those cross-chain features and outputs p(bind), the probability that a given target residue lies in a ligand-binding pocket.
Because the baits are individual amino acids rather than a specific compound, p(bind) flags where ligands tend to bind in general, not the affinity of any particular molecule.
p(bind) is a ranking signal, not a calibrated probability with a universal cutoff. The pocket reveals itself as a tight group of high-scoring residues that sit close together in 3D, not as isolated high values scattered across the sequence.
A practical reading approach:
p(bind) and inspect the top 10 to 15 residues. In the AF2BIND benchmarks, this top slice captures the binding site for most single-pocket proteins.output.pdb. Residues that cluster spatially are the predicted pocket; lone high scorers far from the cluster are usually noise.For proteins with more than one pocket, expect more than one spatial cluster. The ranking alone will not separate them, so the structural view matters.
AF2BIND is strongest when a structure exists and the goal is to localize a small-molecule pocket quickly, especially for a protein where no bound ligand has been resolved. It needs no MSA and no docking box.
If the structure itself is uncertain, predict it first with AlphaFold 2, then score the model with AF2BIND.
Rank a compound library against one protein target with SPRINT protein and ligand co-embeddings and native cosine similarity.
Deep learning framework for predicting small molecule-RNA interactions using RNA secondary structure. Combines language models, CNNs, and graph attention networks for binding prediction.
Predict ADMET (Absorption, Distribution, Metabolism, Excretion, Toxicity) properties from SMILES strings using machine learning models trained on Therapeutics Data Commons datasets.
Predict 22 ADMET properties from SMILES strings with the native Admetica Chemprop models from Datagrok.
Identify toxic, reactive, and pharmacokinetically problematic molecular fragments using structural alert patterns
Predict toxicity and synthetic accessibility of small molecules using machine learning. eToxPred combines toxicity risk assessment with synthetic accessibility scoring to help prioritize drug candidates.
Screen for lead-like compounds using stricter molecular descriptor criteria than Lipinski or Veber rules for early-stage drug discovery
Screen compounds for Pan-Assay Interference patterns that cause false positives in biological assays
Quantitative estimate for protein-protein interaction inhibitor potential. Evaluates drug-likeness for compounds targeting PPIs.
Screen compounds for structural toxicity alerts using PAINS, Brenk, and NIH filters. For focused screening, see PAINS Filter, Brenk Filter, or Veber's Rule.
